
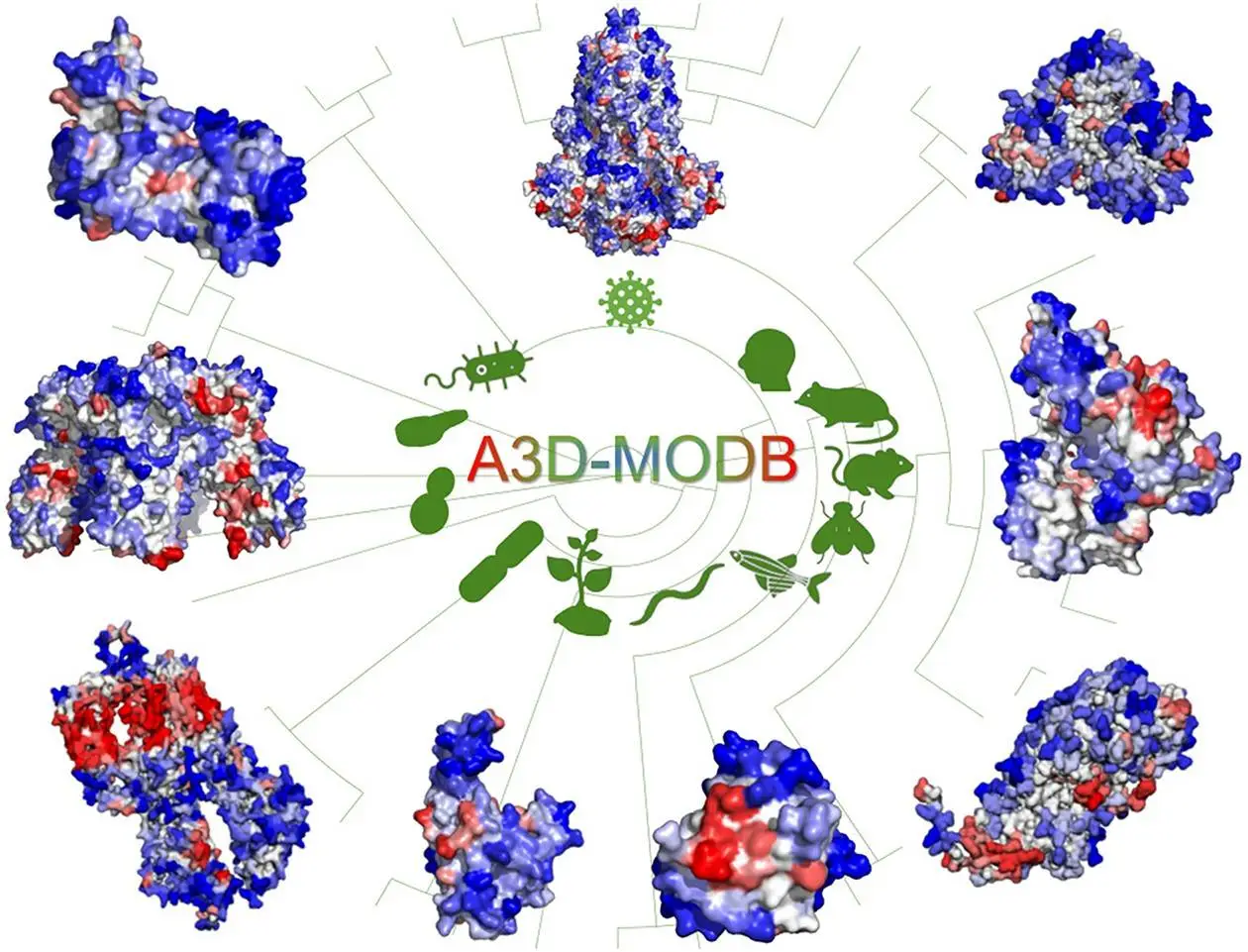
The Protein Folding and Computational Diseases Group at IBB-UAB, in collaboration with researchers from the University of Warsaw, developed a new database, A3D-MOBD (A3D Model Organism Database), that collects information regarding proteomes from 12 of the key model organisms for biological research as well as protein structure predictions to form the most comprehensive resource on protein aggregation to date.
Protein aggregation has previously been linked to aging as well as neurodegenerative disorders like Alzheimer’s disease and Parkinson’s disease. It’s also the cause of increased production costs for several molecules due to obstacles in important purification processes, leading to a lack of accessibility for certain sections of society. Decades of prior research on the proteomes of organisms like Escherichia coli and Saccharomyces cerevisiae has led to the ability to predict sites of protein aggregation computationally.
Tools for computational prediction were remarkably successful, especially with intrinsically disordered proteins, where the prediction could be achieved mainly through sequence analysis due to its significant impact on aggregational tendency. For globular proteins, however, such methods had significant limitations that prevented their widespread use – firstly, they had a tendency to overpredict the influence of collapsed hydrophobic regions on protein aggregation (when they didn’t have much impact) and also predicted mainly on the basis of sequential contiguity, discarding the influence of residues that were sequentially located far apart but would be in close proximity when viewed spatially.
Tools that incorporated information about spatial locations were released, which were able to provide more accurate predictions. Aggrescan 3D was one such tool, which featured a dynamic mode that captured changes in protein flexibility, along with stability evaluation tools, as well as automated tools that facilitated the engineering of proteins for greater solubility and stability. Despite the advances in protein research that A3D facilitated, the limited number of protein structures available for analysis and study restricted its functionality greatly.
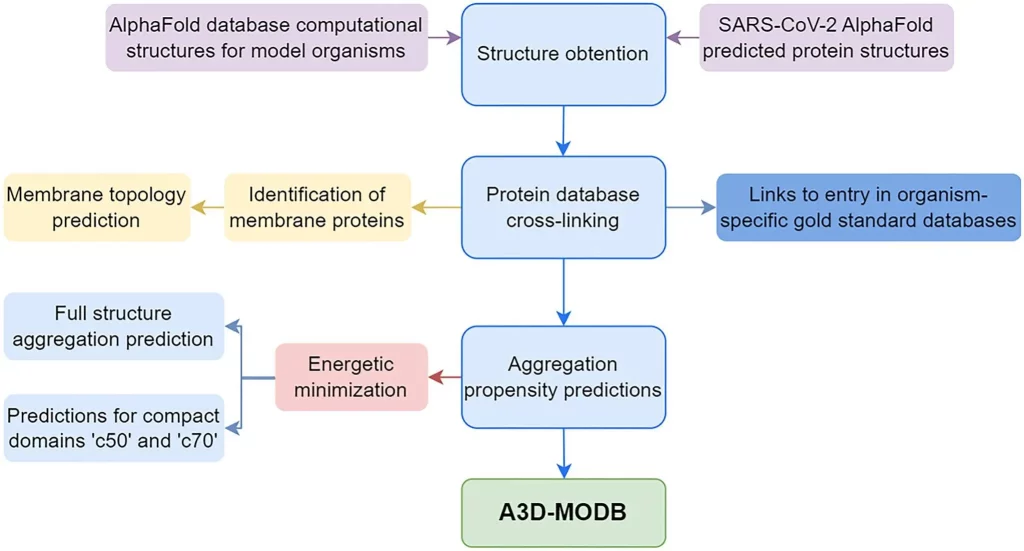
Image Source: https://doi.org/10.1093/nar/gkad942
The release of AlphaFold marked a turning point in the field of protein engineering: nearly every protein ever characterized was computationally analyzed, and highly accurate structures were made available to the public. This data was used to perform analyses of aggregation-prone regions throughout the whole proteome of humans and yeast, and the results of this analysis were made available online, allowing for the study of proteins through the use of large datasets.
Intended as a successor to Aggrescan 3D, a similar method released by the same group in 2015, A3D-MOBD has a greater collection of data available, with almost half a million structure predictions for around 160,000 proteins present in the database. These proteins form the proteomes of several organisms that are used in research across different biological clades. These include, among others, Homo sapiens, Mus musculus, Drosophila melanogaster, and Escherichia coli. A3D-MOBD is constructed using an adaptable architecture that can easily be modified to account for the addition of more data and organisms to the resource.
Additionally, results were given for the stability and solubility of proteins, along with information that provides crucial context to the factors affecting aggregation. Over the course of its development, several tools and computational resources were utilized, such as AlphaFold and TOPCONS, along with organism-specific databases like Wormbase as well as the Human Protein Atlas.
The selection of organisms for the database was made on the basis of clade diversity as well as the frequency of submissions made to A3D. Every protein model available on AlphaFold for a particular species was downloaded and compiled. The A3D tool was used to perform aggregation analysis on the models obtained, which were then uploaded onto the database. The database was also integrated with MySQL, as well as various molecular visualization and data visualization tools, to help users interpret the data in an intuitive and efficient manner. It also incorporates an application program interface that enables programmatic access to the data to make it more suitable for more extensive computational projects.
At present, more than 7000 people have accessed the human proteome database. The database is, at present, the most comprehensive tool for the study of protein aggregation available to the public. Considering the role that the phenomenon plays in the evolution and function of proteins, it is expected that the field will be the subject of increasing interest as protein engineering gains more visibility in academia and industry.
Conclusion
The release of a database centered around this phenomenon may help in providing visibility and aid to researchers in the field, helping to facilitate new discoveries and inventions in space. Certain limitations still exist with the tools, such as the lack of incorporating environmental factors such as temperature and pH on protein structure and conformation. This results in the loss of vital information that may have significant impacts on the aggregation tendencies of various protein regions, resulting in inaccurate predictions. The development of more methods to characterize this impact is crucial to achieving a more comprehensive resource for the study of protein aggregation. Despite this, A3D-MODB’s easily adaptable and accessible framework will allow it to accommodate more resources on various organisms that are relevant in fields like medical technology, agriculture, and genetics research.
Article Source: Reference Paper | Reference Article
Learn More:




Sonal Keni is a consulting scientific writing intern at CBIRT. She is pursuing a BTech in Biotechnology from the Manipal Institute of Technology. Her academic journey has been driven by a profound fascination for the intricate world of biology, and she is particularly drawn to computational biology and oncology. She also enjoys reading and painting in her free time.





{kind=link}