
Protein design is an upcoming niche with important implications for many fields in the realms of healthcare, genetic engineering, and drug creation, among others. However, current methods for sequence design have many disadvantages, including a lack of appropriate accuracy and efficiency. A new model, LigandMPNN, seeks to change this.
The design of protein sequences is important for the creation of small molecule and enzyme sensors and binders, and several tools have been developed to aid in this, including both physical-based methods like Rosetta as well as models utilizing deep learning like ProteinMPNN and IF-ESM. While the latter have been demonstrated to perform better than physical-based methods, they are still limited in their utility due to their inability to adequately integrate non-protein components. ProteinMPNN, for example, only incorporates coordinates of the protein backbone and ignores all other atomic contexts. However, it is important for the design of enzymes, protein-nucleic acid, protein-metal, and protein-molecule binders.
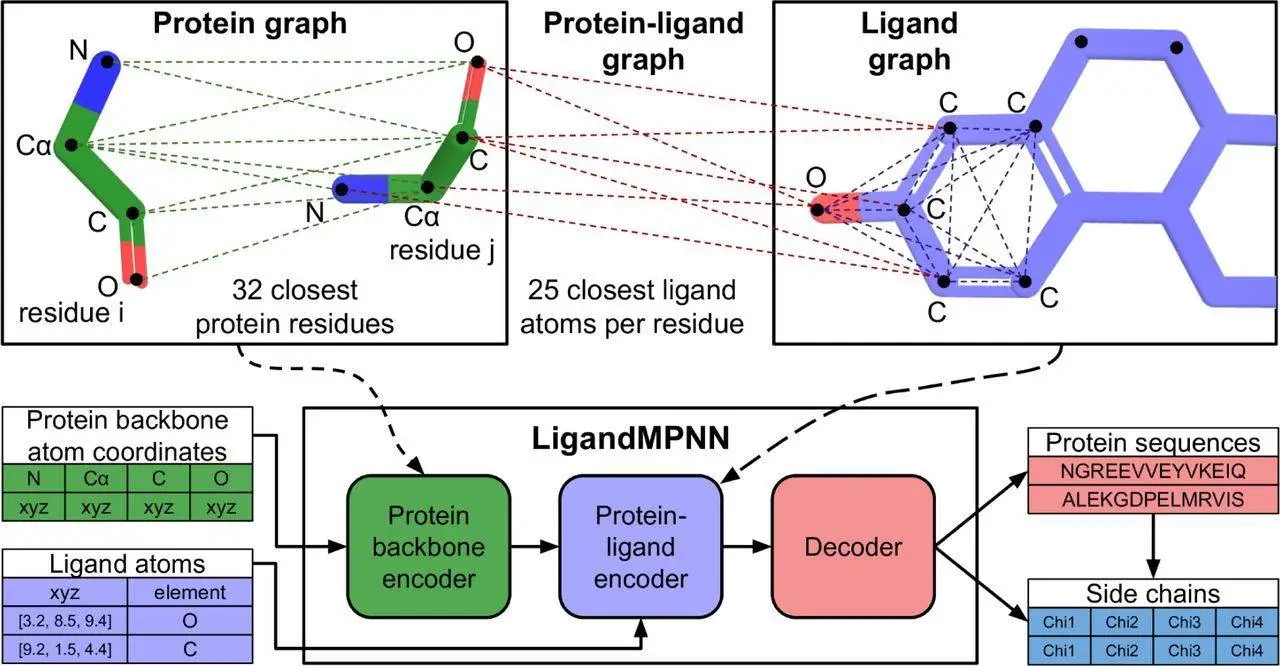
In order to facilitate the design of such binders and sensors, it is necessary to develop a model that takes into account non-protein atoms and molecules and uses it to contextualize the sequence design. This is possible through generalizing the architecture of ProteinMPNN to allow it to take into account non-protein components. Protein residues are treated as nodes and nearest neighbor edges are introduced so a sparse protein graph is created; the geometry of the protein backbone is incorporated into the graph. These inputs are processed to obtain intermediate representations of nodes/edges. As only the closest atoms would materially affect the identity of a given protein residue and its side chain conformations, additional protein-ligand encoder layers were introduced.
A protein-ligand graph was constructed to transfer information to protein residues from ligand atoms. A fully connected ligand graph is also built for every protein residue, and information richness is increased through message passing between ligands. The best performance was obtained through the selection of 25 of the closest ligand atoms for protein-ligand and individual residue graphs. The nodes are initialized to encode chemical elements, and the graph edges encode the distances between them. The protein-ligand encode consists of two message-passing blocks that would modify the graph representations, and its output was combined with the node representations and passed into decoder layers. This model is called LigandMPNN.
In order to assist with the design of multi-state and symmetric proteins, a random autoregressive decoding scheme was used to decode amino acid sequences. Due to the addition of the extra protein-ligand encoder layers and ligand atom geometry encoding, the architecture of the neural network has more than 2.6 million parameters, a significant increase compared to the 1.6 million parameters utilized by Protein MPNN. Both networks are highly efficient, and the time necessary for computation scales linearly according to protein length. The training dataset was augmented with the selection of a random fraction of protein residues and utilizing the atoms comprising their side chains as context ligand atoms. This augmentation did not increase total sequence recoveries but did enable the input of side-chain atom coordinates to the model so functional sites of interest could be stabilized.
A side-chain packing neural network was also trained using the basic architecture of LigandMPNN so as to predict the four side-chain torsion angles for every residue, given amino acid sequence and protein backbone inputs. Three mixing coefficients, three variances, and three means are predicted for each chi angle, of which there are four for each residue. The joint chi-angle distribution is autoregressively decomposed through the decoding of all angles.
The model was trained on the protein assemblies present in the Protein Data Bank, as determined by cryoEM or X-ray crystallography, with a resolution of more than 3.5 Å and a length below 6000 residues. LigandMPNN predicts probability distributions for amino acids as well as uncertainties for every residue position, and the expected confidence correlates with the accuracy of sequence recovery.
When validated experimentally, LigandMPNN was found to redesign and improve binding affinity on binding sites for small molecules, using previously generated outputs from Rosetta that were found to not bind well or at all to their targets. For example, rocuronium, a muscle relaxant, was not known to have any binding associations, as well as cholic acid, whose binding was quite weak – sequences were generated around the ligand using ligand coordinates and backbone as the input, and these retained and introduced new bonding interactions with the sidechain-ligand hydrogen bonding interactions. LigandMPNN managed to increase the binding affinity of cholic acid by more than 100-fold.
Conclusion
LigandMPNN is a deep learning-based approach created for the purpose of designing amino acids that can interact with non-protein atoms and molecules. It is demonstrably superior to physically based methods like Rosetta while being more than 250 times quicker. As no customizations are needed for ligands, this approach is significantly more accessible and easy to use. As LigandMPNN is based primarily on PDB data, some caution is required when using it to design binders for compounds that contain elements that occur rarely in PDB. In a scenario where low data is available, a combination of deep learning-based and physically-based methods may work best.
LigandMPNN’s high accuracy and efficiency make it a preferred method for the design of proteins, and considerable validation has been done to verify its usefulness and applicability. More than 100 DNA-protein and small molecule-protein binding interfaces have been designed using LigandMPNN and have been demonstrated experimentally to be able to bind to their intended targets, thus demonstrating LigandMPNN’s accuracy and utility.
Article Source: Reference Paper | LigandMPNN code available at GitHub
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Sonal Keni is a consulting scientific writing intern at CBIRT. She is pursuing a BTech in Biotechnology from the Manipal Institute of Technology. Her academic journey has been driven by a profound fascination for the intricate world of biology, and she is particularly drawn to computational biology and oncology. She also enjoys reading and painting in her free time.











{kind=link}