
The launch of AlphaFold signaled a revolution in computational modeling: armed with new knowledge of protein structures and unencumbered by limitations on resources and time, scientists were able to use the tool to further their understanding of protein structure, functionality, and interactions. However, AlphaFold’s success in the realm of antibody-antigen modeling is limited. A new study from the University of Maryland evaluates its accuracy and provides new insights into the factors influencing protein modeling.
Antibodies are an important part of the immune system and defend the body from a host of pathogenic organisms due to their high specificity and affinity to antigens. Usually, these targets are engaged through the use of hypervariable CDR loops present in the variable domain. The high specificity of these interactions makes the development of antibodies an important consideration in fields like therapeutics and vaccine development.
The high-resolution structure development of antibody-antigen complexes has served to significantly enhance our understanding of immunity and its underlying mechanisms, allowing for the design of immunogens and antibodies. However, experimental methods for structure determination pose many challenges and constraints on resources and time, often making it infeasible to obtain experimentally derived characterizations for most complexes. In order to fill this need, an array of computational tools have been created – general methods for protein-protein docking have been utilized but haven’t been very successful due to their failure to consider the mobile nature of important CDR loops. Hence, tools have been developed specifically to model antibody-antigen complexes, but obtaining accurate predictions remains challenging.
The introduction of AlphaFold was a significant breakthrough – utilizing deep neural networks to produce accurate structural predictions from the protein sequence, AlphaFold was able to achieve unprecedented accuracy upon its release. An updated version of AlphaFold, AlphaFold-Multimer, was developed specifically for predicting the structures of protein-protein complexes.
Prior benchmarking showed that despite generally being successful in predicting the structures of protein-protein complexes, AlphaFold wasn’t as accurate at modeling antibody-antigen complexes, a limitation that had also been noted by AlphaFold Multimer’s developers. Investigating the factors that influenced failures and successes in modeling can result in valuable insights regarding the protein modeling process and can reveal obstacles and roadblocks, which, when rectified, would result in improvement in accuracy.
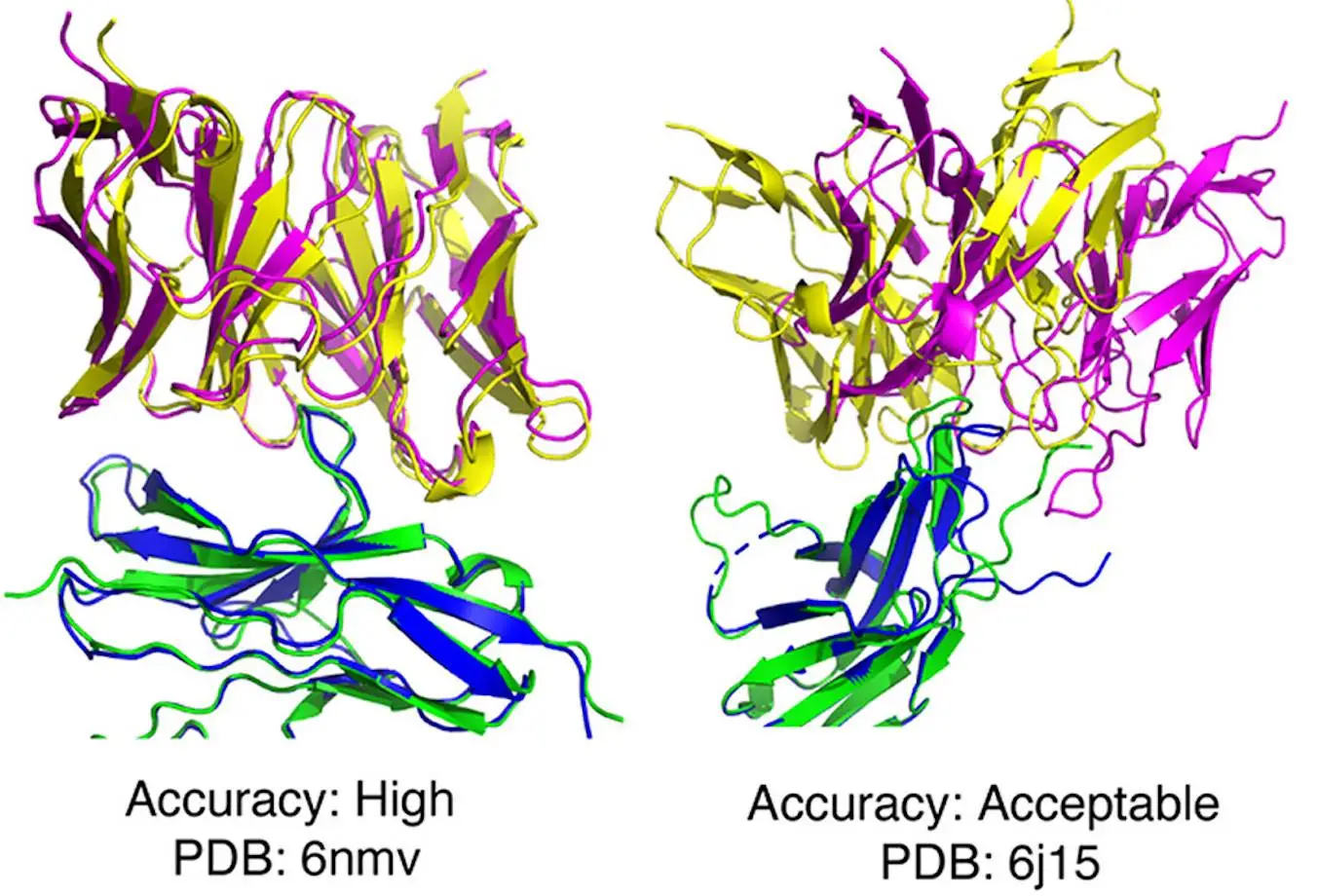
In order to conduct a detailed and comprehensive assessment of AlphaFold’s capacity to model antibody-antigen complexes, a set of more than 400 such complexes was formed. The accuracy of AlphaFold’s predictions was evaluated using CAPRI criteria. AlphaFold was found to generate top-ranked models of acceptable or higher accuracy for 26% of the test cases, medium or higher accuracy for 18% of the test cases, and high accuracy for only 5%. These results were compared to those obtained through other pipelines, like ColabFold. The accuracy of ColabFold was found to be slightly lower, possibly due to the influence of differing structural templates and MSAs. AlphaFold was found to exhibit greater success than algorithms like ZDOCK and ClusPro. AlphaFold v2.2 was found to perform much better than its predecessor, AlphaFold v 2.1.
In order to identify the factors that influence the outcomes of the modeling, the characteristics of the complexes were analyzed and compared to the success of predictive modeling attempts. Analysis revealed that glycans and non-protein components being present at the complex interface was significantly correlated with lower success in terms of predictive modeling.
When evaluating its performance in modeling complexes with heavy-light chains compared to complexes containing nanobodies, no significant statistical difference was found, suggesting that factors like smaller search spaces and fewer CDS loops result in less complexity and size, thus facilitating greater modeling success, it was also found that interactions that were energetically favorable were shown to have better success in terms of modeling.
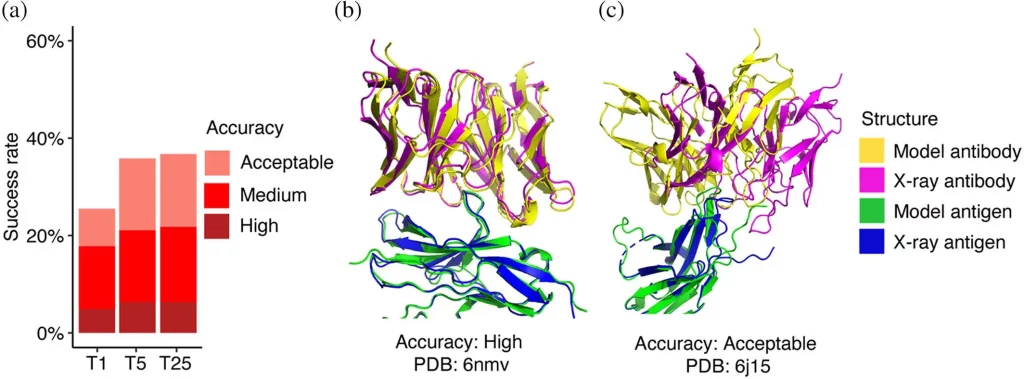
Image Source: https://doi.org/10.1002/pro.4865
Different model accuracy scores were compared in order to assess their ability to correctly discriminate between inaccurate and accurate predictions. AlphaFold’s score, a combination of pTM and iPTM (components of the model confidence scores), along with I-pLDDT, was evaluated, and the latter was found to be superior. The I-pLDDT method also discriminated accurately between incorrect models and models of medium or higher accuracy. I-pLDDT’s greater accuracy may be due to the fact that it is focused on the interface of the antibody-antigen complex, in contrast to ipTM, which is calculated across all inter-chain interfaces of the models, resulting in it being influenced by components that may be less relevant. These findings show that I-pLDDT is a better metric by which the quality of AlphaFold models can be evaluated.
One of the most important components of AlphaFold’s algorithm is its use of recycling iterations: every model generated is used as input into the system for optimization. To understand the impact of these iterations on AlphaFold’s modeling success, the AlphaFold pipeline was modified using ColabFold. Analysis of the outputs demonstrated that model accuracy showed a significant increase as the iterations progressed. Changes in the model across iterations were analyzed in order to identify which features underwent significant modifications and which did not. This helped highlight areas of improvement, as well as optimal features.
It was also found that using subunit chains in bound conformations as input facilitated greater success rates and that the inclusion of MSAs substantially improved accuracy.
Conclusion
AlphaFold’s newer iterations substantially improve on the developments made by its predecessors but still fall short when modeling antibody-antigen structures. Through computational analysis, various factors influencing modeling accuracy were identified, including the presence of glycans and non-protein ligands, the presence of MSA data, and the size and complexity of the models. These gaps can be bridged through further optimization or modifications of the AlphaFold model. Studying these gaps can also further scientists’ understanding of the underlying mechanisms that govern antibody-antigen interactions and advance our knowledge of immunity, with significant implications in the fields of therapeutics, vaccine creation, immunotherapy, and more.
Article Source: Reference Paper
Learn More:




Sonal Keni is a consulting scientific writing intern at CBIRT. She is pursuing a BTech in Biotechnology from the Manipal Institute of Technology. Her academic journey has been driven by a profound fascination for the intricate world of biology, and she is particularly drawn to computational biology and oncology. She also enjoys reading and painting in her free time.











{kind=link}