
Recent advances in large language models (LLMs) and transformers have opened new possibilities for modeling protein sequences as language. Carnegie Mellon University researchers have developed a new AI model called GPCR-BERT that leverages advances in natural language processing to uncover new insights into the sequence-function relationships in G protein-coupled receptors (GPCR), which are the target of over one-third of FDA-approved drugs.
By treating protein sequences like language and fine-tuning a pre-trained transformer model, the researchers were able to predict variations in conserved motifs with high accuracy. Analysis of the model highlighted residues critical for conferring selectivity and conformational changes, shedding new light on the higher-order grammar of these therapeutically important proteins. This demonstrates the potential of large language models to advance our fundamental understanding of protein biology and open up new avenues for drug discovery.
Unraveling the Language of Proteins: GPCR Sequences as a Window to Functionality
Proteins perform vital functions in living organisms and are composed of amino acids arranged in specific sequences. A fundamental question in biology is how the 20 amino acids can evolve into thousands of proteins with diverse functions. While experimental techniques have advanced our knowledge, computational methods provide further insights by leveraging vast amounts of genomic data. However, current bioinformatics models cannot fully capture the complex higher-order interactions within sequences that determine protein structure and function.
Artificial intelligence has recently improved protein predictive models like AlphaFold for structure prediction. However, there has been less focus on structure-agnostic models that use sequences to predict higher-order interactions. With the rise of LLMs and transformers for natural language processing, protein sequences can now be considered as “language” and take advantage of these advances.
This study aimed to develop a transformer model called GPCR-BERT to interpret GPCR sequences. The reasons for focusing on GPCRs are:
- They are the target of over one-third of FDA-approved drugs
- They exhibit functional diversity in many biological processes
- They contain conserved motifs like NPxxY, CWxP, and E/DRY with unknown sequence-function relationships
The key questions explored in the study are:
- What is the correlation between variations in GPCR-conserved motifs and other amino acids?
- Can we predict full GPCR sequences from partial information?
- Which amino acids contribute most to conserved motif variations?
By predicting variations in the motifs and analyzing attention weights, the goal was to shed light on sequence-function relationships and important residues for conformational changes.
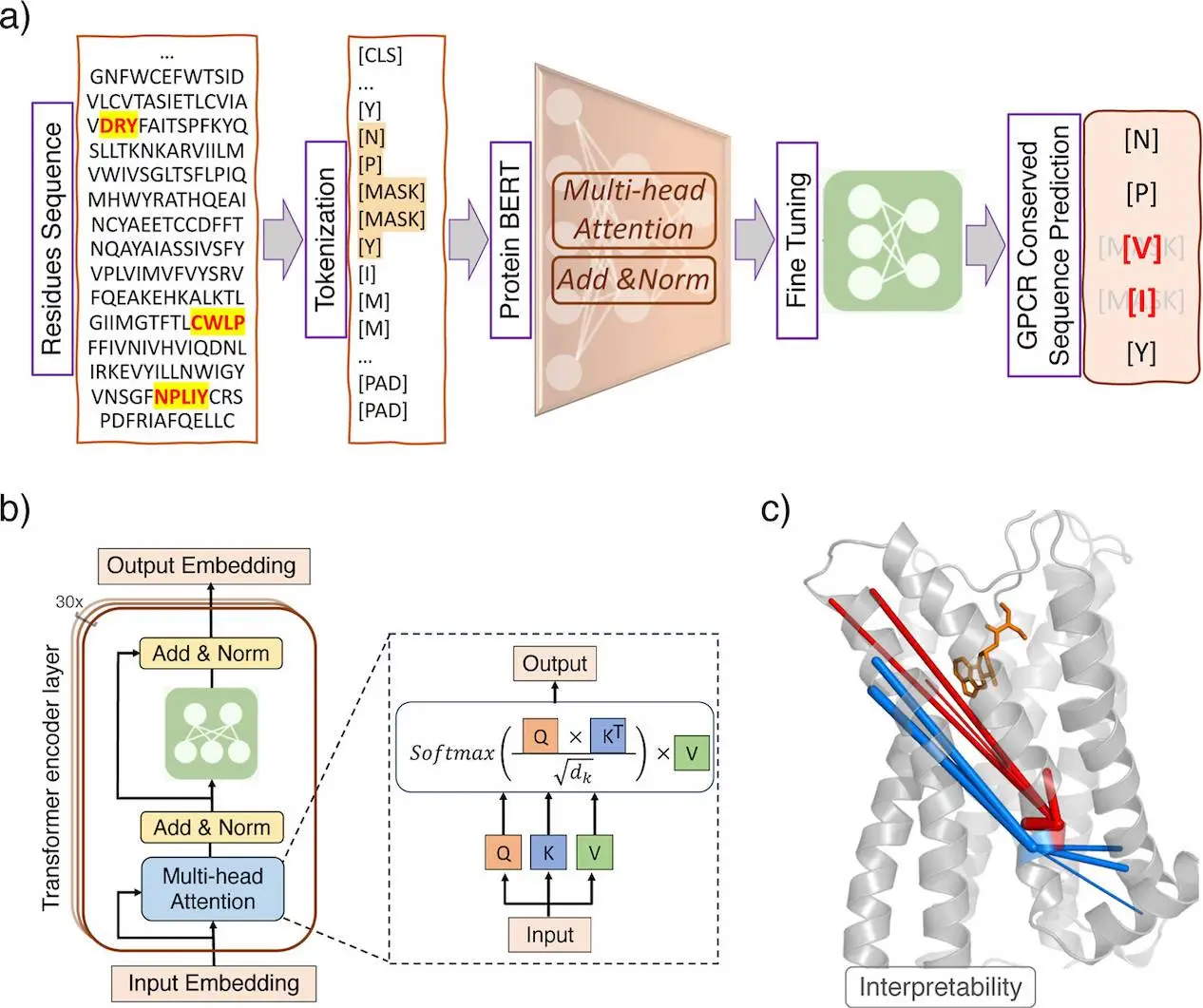
Architecture of GPCR-BERT
The model GPCR-BERT uses Prot-Bert, a pre-trained protein language model, and fine-tunes it for specific tasks on GPCR sequences. Prot-Bert adopts the transformer encoder architecture, which enables modeling long-range dependencies through self-attention.
GPCR sequences containing NPxxY, CWxP, or E/DRY motifs were extracted from the GPCRdb database and tokenized. The residues to predict were labeled as “J” and the model had to predict them from context. Various prediction tasks were defined:
- Predicting xx in NPxxY
- Predicting x in CWxP
- Predicting E/D in E/DRY
- Predicting contiguous masked sequence segments
For each task, Prot-Bert was fine-tuned on the tokenized sequences with a regression head added. The loss function was a cross-entropy loss, and performance was evaluated using multiclass accuracy.
GPCR-BERT’s Performance and Insightful Analysis
The models showed high accuracy in predicting the conserved motif variations, especially for NPxxY and E/DRY. Performance was lower for CWxP, likely due to the smaller dataset size.
The model was also able to predict reasonably well when contiguous segments of the sequence were masked. This suggests that GPCR-BERT can infer complete sequences from partial information, not just conserved motifs.
Compared to BERT and SVM (support vector machine), GPCR-BERT showed much better performance, indicating the benefits of using a protein-specific pre-trained model. Even SVM outperformed BERT, suggesting the generic language model had difficulty learning protein sequences. Analysis of the attention weights revealed that:
- The xx in NPxxY attends most to adjacent and extracellular residues important for ligand binding
- The x in CWxP and E/D in E/DRY attend more to binding pocket residues
This provides clues into how variations in the motifs may alter interactions with ligands.
Visualizing the learned representations also showed that GPCR-BERT can cluster sequences by receptor type, even differentiating subtypes like β1 and β2 adrenergic receptors. This suggests it captures distinguishing features beyond just the conserved motifs.
Comparing predicted important residues to experimental mutagenesis data showed good agreement, validating the model’s insights. Some understudied residues were also identified as potentially significant.
GPCR-BERT: Key Findings and Implications
This study demonstrated that the GPCR-BERT model can provide useful insights into sequence-function relationships in GPCRs by treating sequences as language and leveraging transformer architectures.
Some key findings are:
- Conserved motif variations are linked to other residues, especially ligand binding and pocket regions
- The model can reasonably reconstruct full sequences from partial data
- Attention analysis surfaces residues critical for conformational changes
- The pre-trained protein language model significantly outperforms generic BERT and SVM
These insights could have implications for guiding mutation studies, protein engineering efforts, and drug development.
However, there are some limitations. The model focuses on the most common motif forms and may not apply to rare variations. The motifs also have some inherent variability, like C in CWxP only being 71% conserved.
Overall, this study opened up promising new possibilities for deep learning and language models to uncover hidden patterns in protein sequences. Further development of these methods could lead to a new paradigm in computational biology and drug discovery.
Conclusion
A new AI model, “GPCR-BERT,” is introduced to decipher the complex sequence-function relationships in G protein-coupled receptors. Using a transformer-based language model called Prot-Bert, the researchers fine-tuned the system to predict variations in conserved motifs of these receptors with high accuracy. By treating sequences as language, the model provided new insights into higher-order interactions and sequence-function relationships in this pharmaceutically important protein class. This approach distinguished receptor classes based solely on sequence, highlighting potentially significant residues for mutagenesis studies. This demonstrates the potential of large language models to advance our fundamental understanding of protein biology, guiding future therapeutic development and protein engineering efforts.
Article source: Reference Paper | Code & Data Availability: GitHub
Follow Us!
Learn More:



Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.











{kind=link}