
Scientists from University College London, UK, study the Machine Learning (ML) approaches that have successfully closed the ever-widening gap between large amounts of protein sequences produced by state-of-the-art sequencing methods and annotations of the structures and functions of proteins. One of the best methods for structure prediction, AlphaFold2 (AF2), has resulted in a 1,000-fold increase in structural data for proteins, a remarkable feat in structural bioinformatics. These superfast methods can find relationships between proteins that could not be found using earlier methods. This means that scientists can more easily discover how different proteins are related to each other, even if they are not very similar.
Proteins play essential roles in every cellular process. Protein structure prediction is critical to understanding protein functions. Scientists have wondered about predicting protein structure solely from the amino acid sequence since the first globular protein structures were determined more than 50 years ago.
Currently, the Protein Data Bank (PDB) reports 100,000 unique protein structures, which were experimentally determined using experimental structural biology techniques like NMR, CryoEM, and X-Ray crystallography. However, the vast number of protein sequences generated, in billions, in current metagenomic experiments greatly outnumber the known structures.
Proteins are made of structural units called domains. Domains are largely known to fold independently and are usually associated with specific functional roles. Hence, understanding protein domain structure and folding are crucial in determining protein functions.
Numerous databases like SCoP, CATH, and ECOD have documented and classified domain structures and fold groups into evolutionary superfamilies. These databases have classified up to 5000 domain structures and about 1300 folds. Studies have revealed that these numbers are saturated, and it is the various combinations of domains that result in a vast plethora of protein functions. Hence, understanding domain combinations and evolution is of utmost importance in figuring out the functions of several uncharacterized proteins.
With the advent of Machine Learning approaches in structural biology, protein structure prediction accuracy has been dramatically improved by AlphaFold (AF) using deep neural networks. Along with deep learning (DL), the method also uses evolutionary, physical, and geometric constraints on protein structures.
AlphaFold performed exceedingly well at the Critical Assessment of Structure Prediction (CASP13). Several research groups attempted to replicate the method due to its unavailability to the scientific community in the beginning. RoseTTAFold and PREFMD released new versions competing with AlphaFold, but AlphaFold2 in CASP14 outperformed every other method. The AlphaFold Protein Structure Database (AFDB) is said to have 214 million putative protein structures available, which span the entire UniProt, as announced by DeepMind. These putative protein structures are also said to be available on the 3D-Beacons platform of the European Bioinformatics Institute (EBI).
The authors review various ML approaches for determining distant evolutionary relationships between proteins based on structure and sequence comparisons.
Sequence-based approaches to find homologs
Homology-based inference (HBI) methods have been used successfully to transfer annotations from labeled to sequence-similar unlabeled proteins. Apart from these, Multiple Sequence Alignments (MSAs) are a storehouse of evolutionary information, which are often used in techniques determining de novo protein structures and protein functions. However, these methods are bottlenecked by slow runtimes and faulty parameters, leading to uninformative MSAs. While advanced computing has greatly resolved runtime issues, the ever-increasing databases of sequences require better and more advanced methods.
The pLMs and embeddings as an alternative
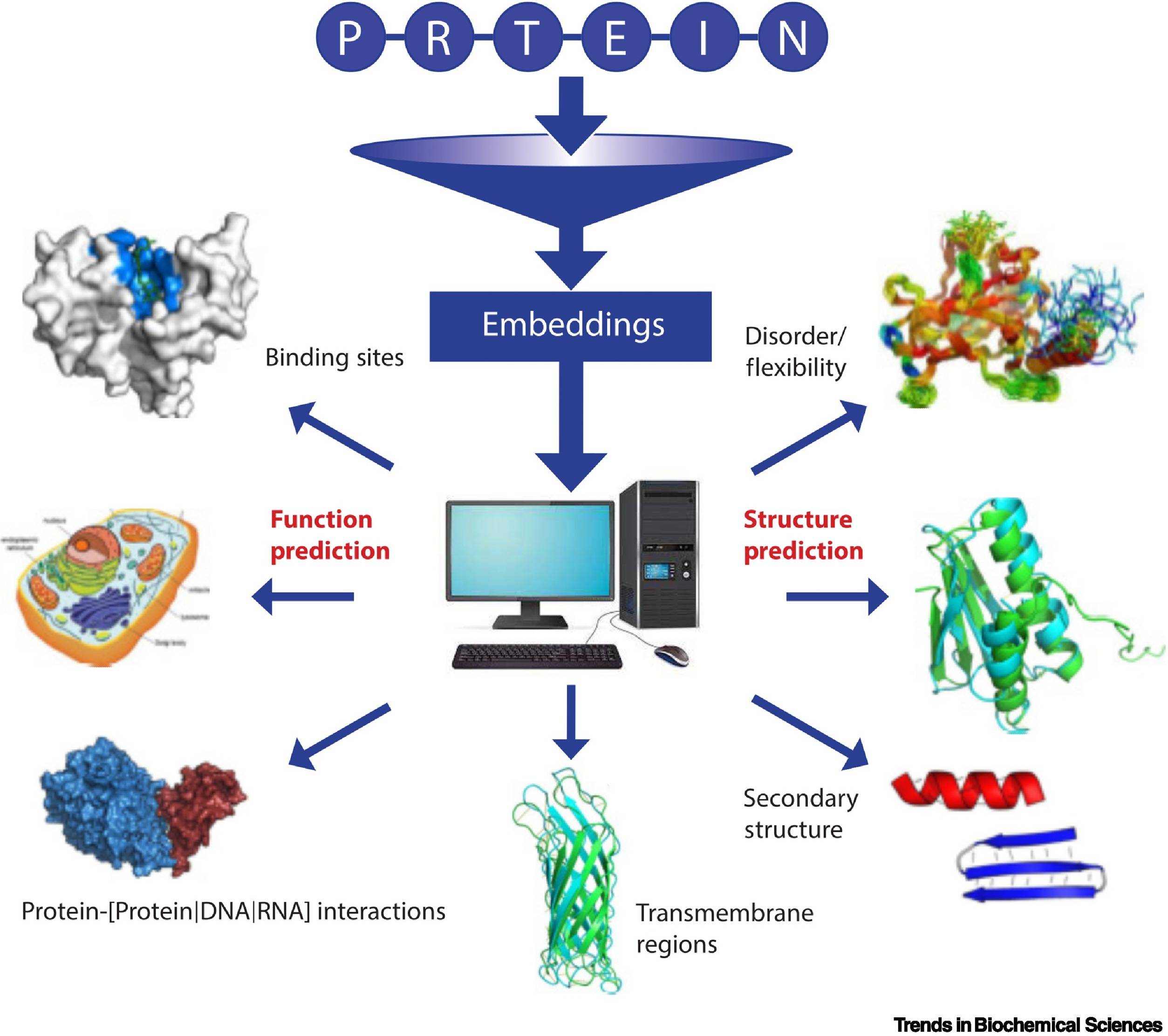
Language models from Natural Language Processing (NLP) prove to be an effective alternative to HBI methods. Incorporating deep learning (DL) techniques to learn the ‘grammar’ of the ‘language’ of life encoded in billions of known protein sequences using the language model setting of NLP. The pLMs (protein language models) learn data attributes such as evolutionary, functional, and structural constraints on protein sequences implicitly, as opposed to typical ML models. Autoregression and masked-language learning are applied to implement the self-supervised learning of pLMs. Autoregression involves training based on predicting a future outcome (in this case, token) based on all previous outcomes (previous tokens). Masked-language learning involves reconstructing corrupted sequences (tokens) from noncorrupted tokens.
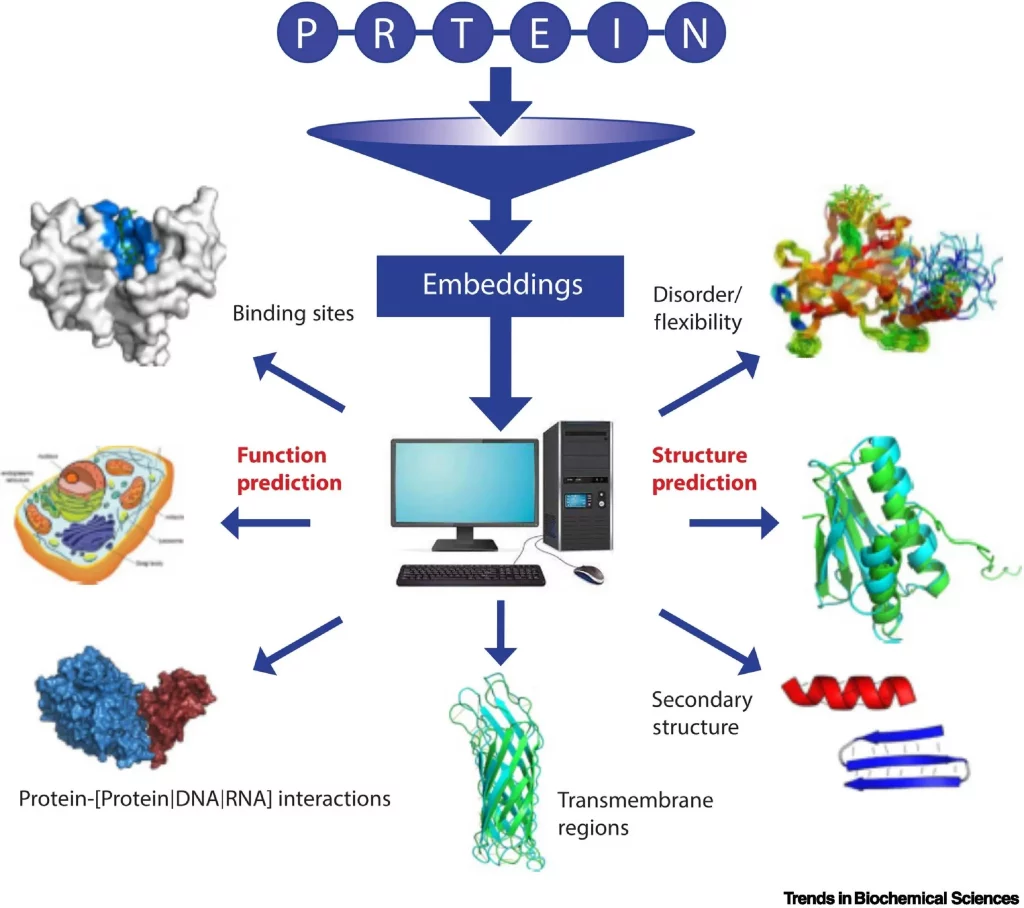
Image source: https://www.cell.com/trends/biochemical-sciences/fulltext/S0968-0004(22)00308-5
Repeating the training with billions of protein sequences results in the pLM learning the statistical properties of the ‘language.’ This can be achieved by extracting embeddings, the hidden states of the neural networks (pLMs). While model training is computationally intensive and typically requires high-performance computing facilities, the extraction of embeddings can be achieved even on PCs or laptops.
The pLMs have been shown to improve over HBI in predicting protein functions. Embedding-based Annotation Transfer (EAT) compares proteins in embedding space as opposed to sequence space for HBI methods. In Critical Assessment of Functional Annotation (CAFA4), EAT outperformed HBI significantly.
Structure-based annotation (SAT) has been shown to capture evolutionarily distant proteins more efficiently than EAT or HBI. Recent breakthroughs in the form of AlphaFold2 in predicting X-ray quality protein structures have led to further applications for SAT methods in structural biology. Studies involving the analysis of protein structures from the AFDB using fold recognition algorithms have resulted in the discovery of novel protein folds.
Traditional Structural aligners have been known to be inefficient in keeping up with the vast number of growing databases of protein structures. Novel structural aligners are emerging using the sequence-based approach, like Foldseek. The pLM embeddings, coupled with fast structural aligners, could span vast regions of the protein fold space in terms of classifying and validating assignments.
The authors analyzed the predicted structural domains in AlphaFold2 for 21 model organisms. The analysis led to identifying 2367 novel families.
The availability of AlphaFold2 as a tool coupled with the AFDB will remarkably speed up research projects which were earlier held back due to a lack of experimentally derived quality protein structures. There are caveats that one needs to keep in mind. Point mutations and single amino acid variants are not addressed. The overrepresentation of protein folding states results in a non-representative model for other alternative states.
Conclusion
Recently, cutting-edge deep-learning techniques have transformed the landscape of protein research. The DL-based method AlphaFold2 dramatically increases the high-quality structural repertoire of proteins with very high accuracy. Using pre-trained language models enables better annotation and prediction than HBI methods. Structural alignment tools coupled with pLMs can traverse the protein space better to predict distant evolutionary relations. In other words, when met with ML approaches, structural bioinformatics methods lead to better, faster, and more advanced methods for protein structure and function determination. These novel ML approaches have the potential to significantly enhance our understanding of proteins and their role in biology and medicine.
Article Source: Reference Paper
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Banhita is a consulting scientific writing intern at CBIRT. She's a mathematician turned bioinformatician. She has gained valuable experience in this field of bioinformatics while working at esteemed institutions like KTH, Sweden, and NCBS, Bangalore. Banhita holds a Master's degree in Mathematics from the prestigious IIT Madras, as well as the University of Western Ontario in Canada. She's is deeply passionate about scientific writing, making her an invaluable asset to any research team.





{kind=link}