
Scientists from UCSD have devised an efficient AI-based pipeline, RESP, for identifying high-affinity antibodies. Typically, researchers would employ directed evolution to enable iterative mutagenesis and selection to identify and assess high-affinity antibodies. Existing methods employing deep learning techniques fail to provide statistical estimates for assessing reliable predictions. RESP, as a pipeline, implements an autoencoder model trained over 3 million B-cell receptor sequences to produce better representations for the input and then implements a Bayesian neural network using ordinal regression to model the relationship between the directed evolution sequence data and the binding affinity information. The method is not restricted by the scope of the search space but rather is able to assess sequences out of the directed evolution sequences library. As a proof of concept, the authors demonstrated a 17-fold increase in the KD PD-L1 antibody Atezolizumab. Fast identification of high-affinity antibodies will also aid in producing monoclonal antibodies, a requirement in therapeutics.
High-affinity antibody drugs
An antibody must bond strongly with its target to be a successful drug. The process of generating such antibodies involves directed evolution strategies in the laboratory. The procedure involves starting with a known antibody amino acid sequence and developing mutated variants of the sequence using bacteria or yeast cells. The mutants are then assessed for their binding capabilities to the specific target, and the best-performing candidates are put through several rounds of mutagenesis and assessment until suitable tight-binding candidates have evolved.
Though antibody development is time-consuming and expensive, they frequently prove ineffective in clinical trials. To make this process quicker and more effective, researchers introduced the machine learning approach, which was used to find a new antibody that binds a key cancer target 17 times more tightly than an existing antibody drug.
Much-needed desideratum for high-affinity antibody identification
Earlier methods relied on free-energy estimation for identifying and assessing antibody binding affinity. These methods were limited owing to their high cost and low throughput. As an alternative approach, various machine learning-based techniques were developed addressing various aspects of antibody design. But these methods have failed to provide confidence intervals and uncertainty estimates for assessing prediction reliability. While Gaussian processes have been suggested as an alternate candidate, they cannot address datasets containing more than 5000 sequences.
The other key challenge is the choice of an appropriate encoding scheme for the inputs. While several processes are already available, there is no consensus in the community regarding the choice of method for a particular problem. The machine learning technique of one-hot encoding, which converts categorical data to the machine-learning algorithm readable form, although simple to implement, is often uninformative and requires unnecessary high-dimensional implementations.
The authors have developed a machine learning-based pipeline, RESP, for identifying high-affinity antibodies. The pipeline is easy to implement and addresses the limitations mentioned above and the challenges of the previous methods, a much-needed desideratum.
Overview of the RESP pipeline
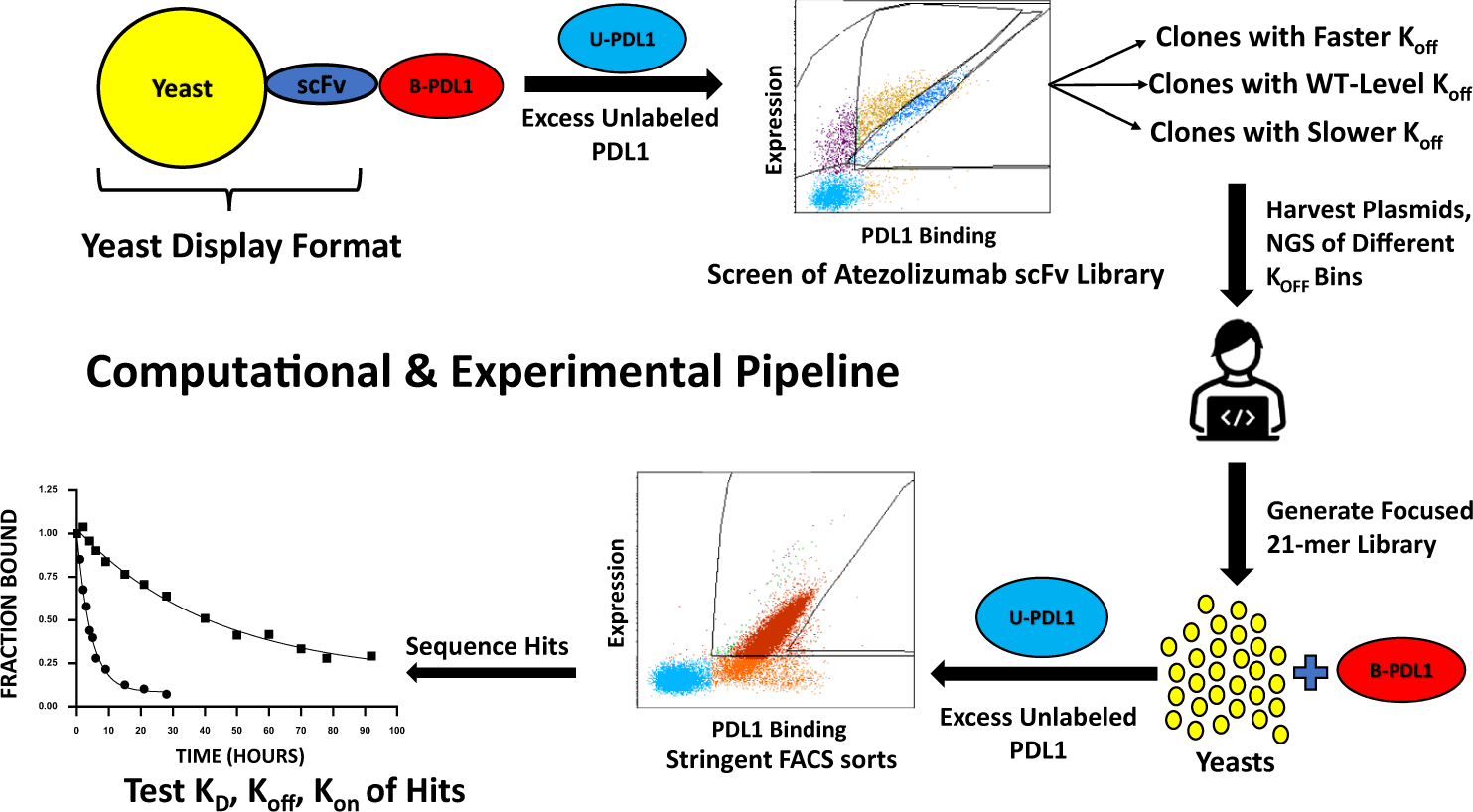
The pipeline has four main components:
- Encoding scheme: A convolutional autoencoder comprising three components is modeled to produce learned representations incorporating distinguishing features of the human B-cell receptor(BCR) sequences from other evolutionarily close sequences. This representation generation is applicable to any antibody sequence of interest and can be used for other projects as well.
- When using the RESPpeline to develop antibodies against a specific antigen, the training set should be target specific. This is because the model will then learn to recognize the sequence type that will bind the specific target. The antigen of choice for the authors is the programmed death ligand (PD-L1), and the antibody is the single chain fragment variable(scFv) heavy chain of the Atezolizumab antibody.
- The next step is the affinity modeling of the sorted sequences from step 2. This step takes a sequence representation as input and predicts the category to which it belongs. The three categories of decreasing off-rates are RH01, RH02, and RH03. This step incorporates antigen-specific experimental data and requires retraining with every new choice of antigen. The model implements ordinal regression to achieve this categorical sorting of sequences and generate a latent score value. The score is an indicator of the binding affinity- the higher the score, the more likely that the sequence should belong to a more stringent sort. The Bayesian neural network, as implemented, is shown to generate an estimate of uncertainty.
- The next step is the in-silico-directed evolution for selecting the most promising candidates for the specific target. The steps involved are illustrated in the following figure.
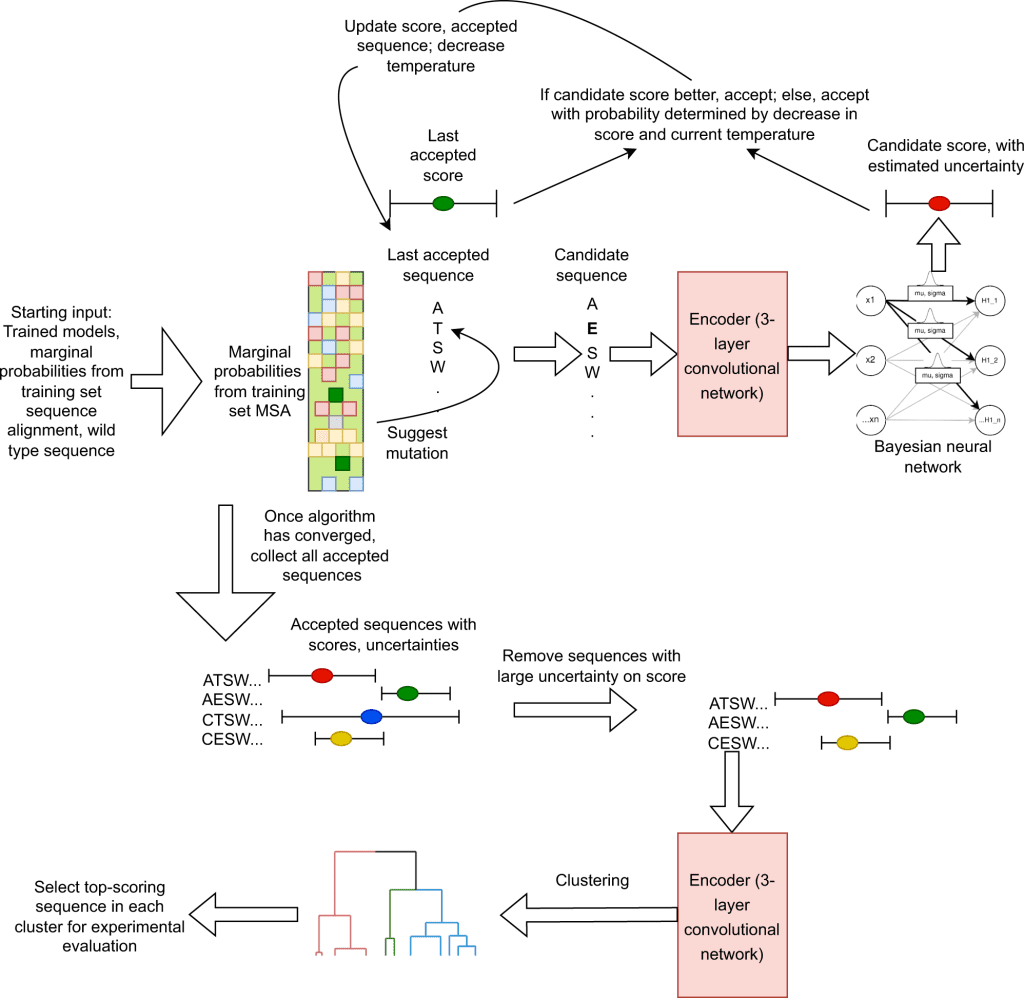
Image source:https://www.nature.com/articles/s41467-023-36028-8/figures/3
The outcome of the study
- The autoencoder was trained on the dataset from the cAb-Rep database, which is fully one-hot encoded. The training involved a decoy dataset with a test set of 200,000 sequences. The model prediction for the B-cell receptor was highly accurate, with 97.4% accuracy. The test had a reconstruction accuracy of 99.99%. The findings suggest that the autoencoder is capable of compressing the input sequence and retaining its properties for its classification.
- The authors on constructing a single library, were able to select a tight-binding antibody. This is purely achieved using computational techniques and avoids experimental techniques for KD measurements.
- The authors compared their method with the previous method by Mason et al. RESP performs better in terms of constraining the search space using the simulated annealing algorithm compared to the Mason et al. study, which generates libraries after several iterations of mutagenesis using a rational design based on data from previous steps. The Bayesian neural network, as compared to the CNN( convolution neural network) proposed by Mason et al., provides uncertainty estimates for better elimination of unreliable predictions.
Conclusion
The authors have developed a novel pipeline, RESP, for developing antibodies that could revolutionize therapeutics involving diseases like cancer, arthritis, COVID-19, and many more. The potential of the method in general antibody development is found to be remarkable, both in terms of efficacy as well as time requirements. RESP provides uncertainty estimates that were not provided by any previous method. This leads to the better generation of predictions for tight binding antibodies for the target antigen and hence promises to produce better and more reliable antigen target-specific antibodies with high binding affinities.
Article Source: Reference Paper | Reference Article
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Banhita is a consulting scientific writing intern at CBIRT. She's a mathematician turned bioinformatician. She has gained valuable experience in this field of bioinformatics while working at esteemed institutions like KTH, Sweden, and NCBS, Bangalore. Banhita holds a Master's degree in Mathematics from the prestigious IIT Madras, as well as the University of Western Ontario in Canada. She's is deeply passionate about scientific writing, making her an invaluable asset to any research team.





{kind=link}