
Scientists from the Broad Institute of MIT and Harvard discovered an approach to studying the functional implications of the millions of mutations linked to cancer by pooling Perturb-seq and measuring the impact of TP53 and KRAS variants on RNA profiles in single cancer cells.
Precision medicine entails predicting how specific genetic variants affect a patient’s health. However, even within recognized cancer genes, most reported variants are uncommon, and because recurrence might indicate either functional selection or random background mutational processes, assessing variant effect remains a difficulty for deciphering cancer genomes. Previous research has employed computational or experimental methods to anticipate the possible functional impact of variants, which are characterized as significant differences from the wildtype (WT) allele.
The researchers used single-cell RNA-sequencing (scRNA-seq) readout with Perturb-sequencing for pooled genetic screens to profile coding variants utilizing single-cell expression-based variant effect phenotyping (sc-eVIP). They looked at 200 TP53, and KRAS gene variants and created computational methods for determining the functional impact of individual variants. They also showed how population-based assessments could miss the effects of variants on single-cell heterogeneity.
Perturb-seq was adapted to concurrently resolve the identity of an exogenously introduced cancer variant marked with a DNA barcode and the induced expression state at the single-cell level to analyze the impact of coding variants at scale. “Variant function could be determined by comparing expression profiles between cells with variant and Wild Type (WT) gene constructs,” the researchers reasoned. To allow pooling, the coding sequence of each variant was cloned onto a modified Perturb-seq vector, tagged with a unique DNA barcode, and 3′ scRNA-seq was used to extract both the expression profile of each cell and the variant(s) it overexpressed.
As a test case, the researchers looked at 100 coding variants in TP53, a tumor suppressor gene that is frequently mutated in cancer. They then transduced the variants into cells at a low multiplicity of infection (MOI) to favor single variants per cell, selected for successfully infected cells for two days using puromycin, and allowed the cells to recover for another two days after that performed scRNA-seq. The researchers recovered 162,314 high-quality cells, with 84% having identifiable variant barcodes and 62% having a single variation annotation. More than one barcode unique molecular identifier (UMIs) was used to support >70% of variant assignments to cells. Only two TP53 variants exceeded a 1.5-fold expression differential from the WT construct, with most variants expressing at levels equivalent to the WT construct. They only used cells harboring a single variant for analysis.
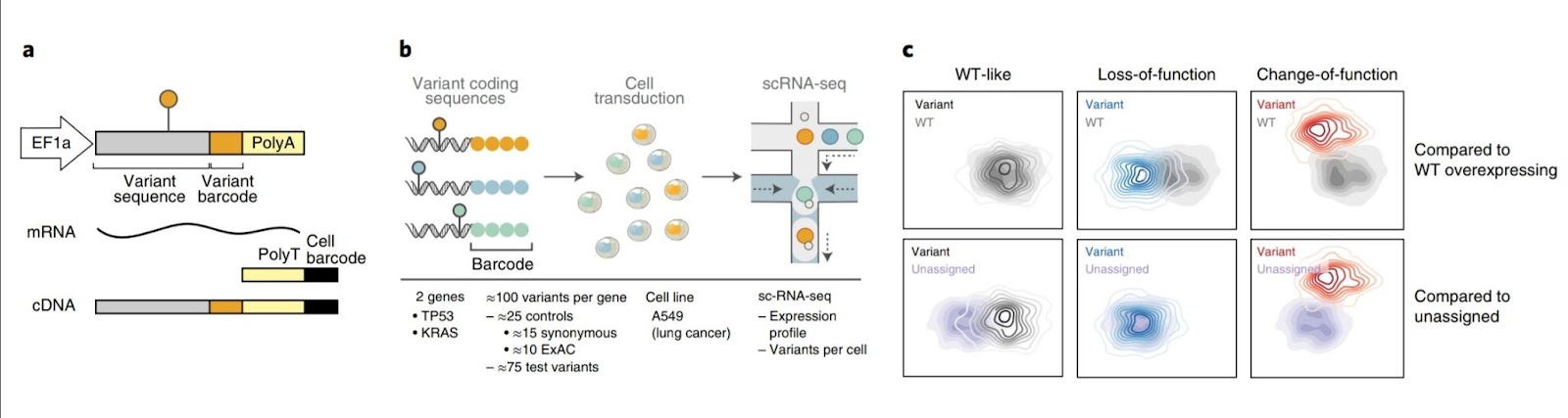
Image source:Massively parallel phenotyping of coding variants in cancer with Perturb-seq
Functional TP53 variants are identified using variant impact scores. The researchers classified variants based on their expression profiles, using two methods: (1) assessing the distance between variant and control construct profiles and (2) unsupervised clustering of variants. When compared to an empirical null distribution produced from comparisons between control synonymous variants, these variants were labeled as ‘impactful.’ Expected loss-of-function/dominant negative variants were differentiated from control variants using both sc-eVIP scores and clustering. All synonymous and ExAC control variants formed a distinct cluster (‘WT-like’) with sc-eVIP scores similar to WT. These results show that the variants in this cluster were accurately classed as loss-of-function because they comprised cells without an identified variant (referred to as ‘unassigned’ cells).
Both sc-eVIP scores and expression subclusters were used to divide the variants that were not in the WT-like cluster into two categories (Impactful I and II). Impactful I variants induced canonical TP53 signatures to a lesser extent than WT-like variants, whereas Impactful II variants had no effect (or repressed) on them.
Compared to unassigned cells, dominant negative and WT-like variants had diametrically opposed effects on multiple gene programs. Using expression profiles to predict whether any individual cell carries an impactful or WT-like variant had limited accuracy. In contrast, overall variant classification using the proportions of cells in each cell cycle phase was highly accurate, implying that TP53 variant phenotypes are primarily due to a shift in cell state distributions. These proof-of-concept results validate the Perturb-seq approach for highly accurate functional classification of TP53 variants.
Although A549 cells have the KRAS G12S allele, it was previously discovered that in cellular tests, they allow for reliable distinction of the function of exogenously produced KRAS alleles.
The researchers chose the 75 most common KRAS alleles and 26 negative controls. At some hotspot locations, both sc-eVIP scores and grouping by mean expression profiles differentiated synonymous KRAS alleles from known gain-of-function variants. Clustering identified another 19 variants as WT-like, including nine out of ten ExAC control variants. An orthogonal KRAS cellular assay evaluating growth in a low attachment (GILA) in human embryonic kidney cells (HA1E) overexpressing KRAS alleles had a high association with sc-eVIP scores. Furthermore, the most frequently mutated variants in cancer cohorts were among those with the highest sc-eVIP scores. Overall, these results back up the accuracy of our single-cell categorization method.
Next, Principal component analysis (PCA) was used to look for principal components that were considerably higher in cells bearing gain-of-function variants compared to synonymous variants. Individual cells with activating vs. WT-like variants were best differentiated by principal component 3 (PC3) scores. PC3 scores were comparable to WT-overexpressing cells, additional variants were categorized on a scale of increasing PC3 scores. They progressed from the mildest separation in WT-like cluster variants with significant sc-eVIP scores to the strongest separation in gain-of-function variants. A similar pattern in PC4 shows that there may be two sources of variants at work.
The researchers then analyzed the distribution of single-cell profiles across variants in a 2D embedding to see if the observed continuum develops because mutations produce a new cell state, redistribute in the same phenotypic space, or both. WT-like and Impactful variants occupied a mostly overlapping cell state space. Still, there was a continuous shift in cell distribution across this space, from WT-like to Impactful I, II, and III, and then the continuum of gain-of-function variants. In all but two variant classes, a model based on mean principal component scores per variant performs near-perfectly, implying that variants have an impact on cell composition. These results indicate that KRAS variants may exert their effects by shifting distributions at the single-cell level.
Models trained using cellular assays on a more extensive dataset performed best, implying that estimates of mutability and effect can become more predictive as dataset size grows. As evidenced by KRAS, this assay is especially useful for uncommon mutations. In contrast, some, but not all, cancer cohort mutations with lower sc-eVIP scores (in the WT-like and Impactful I groups) show higher mutability. Finally, predictive models based on either sc-eVIP or GILA scores performed similarly to previous models. These findings emphasize the use of functional data in addition to cancer genome sequencing.
After assessing the approach’s scalability, large variant effects can be successfully detected with 20–100 cells per variant, but minor effects necessitate 100–400 cells. According to this downsampling calculation, 8.3 million cells could study all 270,000 possible variants in each 1 kb cancer gene in the Foundation Medicine Panel, and 71 million cells could create a draught cancer variant functional atlas of roughly 2.3 million possible variants in the approximately 200 actionable cancer genes under 3 kb. Such comprehensive tests now look within reach, as large-scale single-cell profiling and DNA synthesis technologies scale up at decreasing prices.
Researchers anticipate variants in coding Perturb-seq combined with the sc-eVIP analytical framework offers a flexible way to phenotyping variant’s impact. It can be used right away to evaluate medium-sized libraries of hundreds of disease-related variants. With further refinement, it should be able to handle tens of thousands of variants in cancer and beyond and build general predictive models linking genetic variants to cellular functions. A foundational resource for translational cancer research would be a publicly accessible atlas of variant impact across key cancer genes and contexts.
Story Source: Ursu, O., Neal, J.T., Shea, E. et al. Massively parallel phenotyping of coding variants in cancer with Perturb-seq. Nat Biotechnol (2022). https://doi.org/10.1038/s41587-021-01160-7
Background Image Source: https://www.broadinstitute.org/news/new-high-throughput-method-greatly-expands-view-how-mutations-impact-cells
Data and code availability : https://doi.org/10.1038/s41587-021-01160-7
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Swati Yadav is a consulting intern at CBIRT. She is currently pursuing her Master's from Delhi Technological University (DTU), Delhi. She is a technology enthusiast and has a keen interest in the latest research on bioinformatics and biotechnology. She is passionate about exploring new advancements in biosciences and their real-life application.











{kind=link}