
The researchers from MIT Cambridge, USA, and collaborators at the University of Pittsburgh School of Medicine, present DYNAMO – an analytical framework that infers absolute RNA velocity, reconstruct continuous vector fields that predict cell fates, uses differential geometry to obtain underlying regulations, and anticipates optimal reprogramming routes and perturbation results.
The capability of a single zygote to differentiate into a variety of cell types while preserving the same genome is a hallmark of metazoans. Waddington used the epigenetic landscape as a metaphor for this process, in which differentiation is equivalent to a ball sliding downhill into numerous valleys. This metaphor has been used to explain cell differentiation, transdifferentiation, and reprogramming intuitively; nonetheless, a fundamental goal of the discipline is to go beyond such a qualitative and metaphorical conception and toward more quantitative, statistical models.
Mathematical modeling, particularly when combined with dynamical systems theories, is a valuable tool for understanding how gene regulatory networks (GRNs) influence biological processes. Despite the efforts to perform whole-cell simulations of bacteria, recreating the vector field that indicates the temporal variations of a genome-wide expression state in mammalian cells from experimental data remains a significant challenge.
Recent advances in single-cell genomics have made it possible to profile cell-state transitions with remarkable precision. The development of computer techniques for implying cellular dynamics from snapshot observations has been fuelled by advances in single-cell profiling. The invention of RNA velocity, which explicitly explores the intrinsic splicing kinetics to anticipate the cell RNA expression states in the future, is the other significant advancement. Several groups have recently applied single-cell techniques to bulk RNA-seq with metabolic labeling. By separating “new” and “old” RNA molecules in an experimentally controlled manner, time-resolved single-cell RNA-seq (scRNA-seq), or transcriptome single-cell RNA-seq (tscRNA-seq), gives additional quantitative metrics of cell state and velocity. In theory, these approaches provide the data required for accurate transcriptomic vector field reconstruction.
Here, The researchers present a methodology for creating and analyzing single-cell transcriptomic vector fields in this work. The framework introduces four new features. To begin, they develop an all-encompassing model of expression dynamics that not only reliably estimates genome-wide RNA turnover rates but also overcomes the inherent limits of traditional splicing-based RNA velocity inferring absolute velocities. Second, from discontinuous, sparse, and stochastic single-cell observations, they create a universal technique for robustly reconstructing the continuous transcriptomic vector field. Third, to acquire additional biological insights, they combine the scalability of machine learning-based vector field reconstruction approaches with the interpretability of differential geometry analysis, such as Jacobian, acceleration, curvature, and divergence. Fourth, they create two principle methodologies, least action pathways (LAPs) and in silico perturbation, to generate non-trivial predictions of optimal paths and significant drivers of cell-fate transitions, as well as genetic perturbation outcomes.
This approach marks a significant step forward, from the metaphor of the epigenetic landscape to a quantitative and predictive theory of single-cell transcriptomic’s temporal evolution, applicable to a wide range of biological processes and at a genome-wide scale.
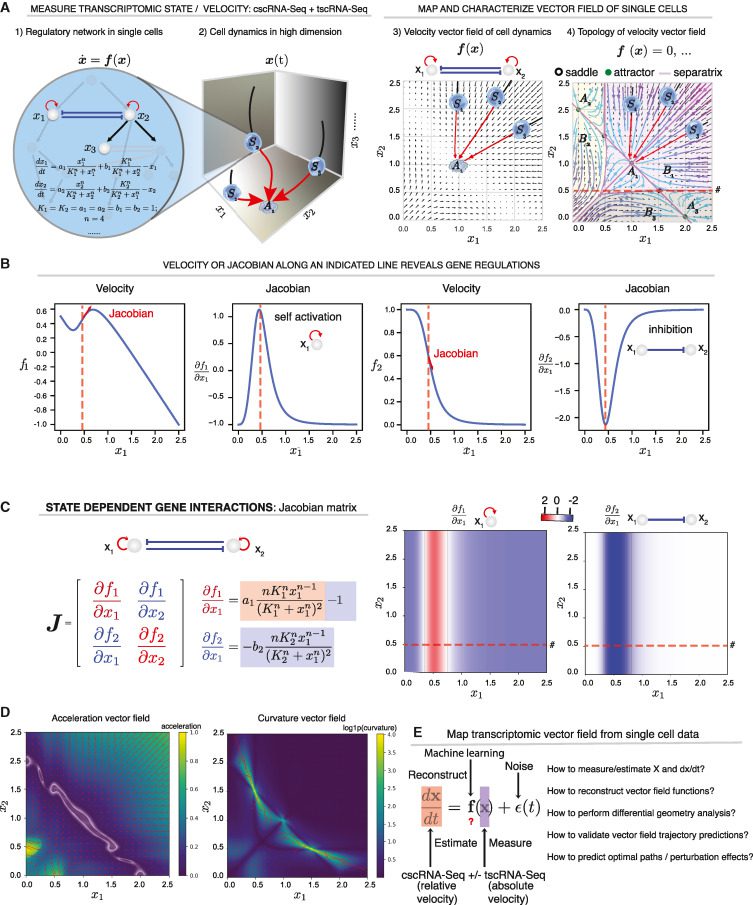
Image Source: Mapping transcriptome vector fields of single cells.
A velocity vector field, in theory, provides a complete account of how genes control one another. Consider the PU.1/SPI1-GATA1 regulatory network involved in hematopoiesis as a simple example of a two-gene toggle-switch pattern that frequently emerges in cell differentiation. The vector field for this pattern is commonly expressed as a series of ordinary differential equations (ODE)s that represent the self-activation and mutual inhibition of PU.1 and GATA1, specify a cell’s instantaneous velocity at any given expression state and anticipate the evolution of the cell state over time. Separatrices (divide the space into three attractor basins, each holding a stable fixed point, i.e., the attractor, corresponding to a stable phenotype), can be used to further define the topology of this vector field in its gene expression space.
Analyzing the vector field can also aid in developing theories regarding how genes control cell states. For instance, the Jacobian can explore the cell state-dependent interactions because it is closely related to the underlying regulatory network.
A variety of other differential geometric variables provide further gene regulatory information. The acceleration field indicates gene expression subspaces (i.e., cell state hotspots) when velocities change drastically in magnitude or direction. Before slowing down in the attractor state, a cell’s velocity tends to increase as it exits an unstable condition and advances toward a stable attractor condition. As a result, detection is possible for genes with a substantial value for acceleration (in magnitude) in unstable states, making essential contributions to cell-fate commitment, long before cells exhibit observable lineage-specific gene expression differences.
The curvature field is a related but distinct quantity that displays gene expression hotspots when the velocity abruptly changes direction. Regulating genes that control cell destiny are the genes that have the most impact on curvature.
Curl and divergence describe the microscopic rotation of a cell state in the vector field, as well as the local flux exiting vs. entering a tiny location in the expression space— the “outgoingness.”
The original RNA velocity approach assumes a universal splicing rate constant and leverages accidentally collected intron reads from cscRNA-seq data. The researchers built an inclusive model that considers RNA metabolic labeling (when utilizing tscRNA-seq data), RNA splicing, and degradation to establish a unified framework for collecting RNA kinetic information from cscRNA-seq and tscRNA-seq datasets. They also use three simplified models to account for different data kinds and experiments: Model 1 is customized for cscRNA-seq and considers RNA transcription, splicing, and degradation but not RNA metabolic labeling, whereas Models 2 and 3 are designed for tscRNA-seq with metabolic labeling, but only Model 3 addresses RNA splicing.
Dynamo can be utilized to calculate the relative degradation rate constant and relative spliced RNA velocity when just cscRNA-seq data are available or when splicing data from tscRNA-seq trials is required. Dynamo addresses the inherent limits of traditional RNA velocity estimation, resulting in inaccurate velocity measurements, allowing for more precise absolute vector field analysis downstream.
Single-cell nucleotide sequencing (scNT-seq) was used to build a time-resolved scRNA-seq dataset using primary human HSPCs (hematopoietic stem cells) and progenitor cells to demonstrate that, large-scale UMI-based tscRNA-seq datasets improve velocity analysis over cscRNA-seq datasets. The labeling data (labeled and total RNA) generated velocity flows that closely matched the established understanding of hematopoiesis using dynamo’s modeling framework.
The transcription rate for labeling data is modeled by Dynamo as a variable that depends on measured fresh RNA and varies across genes and cells.
In the second step, the researchers use single-cell velocity vector samples as input to learn a continuous vector field in transcriptomic space. To understand the transcriptome vector field scalably, efficiently and robustly from noisy and sparse data of single-cell states and velocity estimations, a machine learning strategy was used that takes advantage of recent developments in vector-valued function approximation. Once a vector field is understood, the immediate application is to predict the past or future state of a cell in a way similar to Newton physics, i.e., one may anticipate position and velocity at a certain point in time using the vector field and the initial gene expression states. The researchers reasoned that “this prediction can be confirmed by comparing the single-cell trajectory prediction with gene expression in clonal cells monitored sequentially, which approximates the dynamics of a single cell over time”. This technique may also infer the transcriptome dynamics of cell ensembles across time in silico, which could be a valuable addition to live-cell imaging or lineage tracking.
In the third stage, the researchers constructed a coherent suite of differential geometric studies with the vector field to find quantitative information about gene regulation. To obtain mechanistic insights, these methods were used in their hematopoiesis tscRNA-seq dataset. With this dataset, they learned the vector field for the first time. The topology of the system is accurately reflected by the fixed points indicated in the uniform manifold approximation and projection (UMAP) space-based vector field. Following that, the vector field was arranged into a tree structure that accurately describes the hematopoietic lineage hierarchy. As a result of Dynamo, single-cell genomics data may be used to directly investigate controlling regulatory mechanisms and even retrieve kinetic characteristics underlying cell-fate changes, such as Hill coefficients.
The researchers wanted to establish a principled technique that reveals ideal pathways, associated driving TFs (transcription factors), and the corresponding expression dynamics with them using the continuous vector field built from scRNA-seq datasets. With multiple established developmental, dedifferentiation, and transdifferentiation processes, the hematopoietic scNT-seq dataset is ideal for evaluating such a methodology.
In the fourth stage, two fundamental methodologies were presented for predicting optimal transition pathways and genetic perturbation outcomes: LAPs and in silico perturbation.
The Least Action Path LAP (action: a trajectory functional) is a principled strategy that has previously been utilized in theoretical attempts to estimate the most likely path a cell would take during destiny transition. The researchers reasoned that “using the LAP approach and the analytical vector field, we could generate principled predictions of the best hematopoietic cellular conversions.” The LAP method’s ability to estimate the main transition path and accompanying gene expression patterns broadly provides non-trivial transition predictions. One can project the LAP back to the original gene expression space to predict the entire transcriptome kinetics along the path once it has been calculated in the PCA (principal component analysis) space. “The findings show that the LAP approach has the capacity to accurately anticipate the best route and TF combinations of cell-fate transitions, paving the way for a’la carte reprogramming between any cell types of choice for regenerative medicine applications.” The researchers explained.
The analytical form of a vector field enables in silico perturbation predictions of expression reaction for every gene in each cell, as well as cell-fate dissimilarities following genetic perturbations. They showed the predictive ability of hematopoietic fate trajectory predictions following genetic alterations in particular. Other cellular transitions are likewise correctly predicted by in silico perturbation. The capacity to perform in silico perturbations should aid in the search for gene combinations that rise to intriguing cell states and transitions among the huge number of possible pairwise and higher-order perturbations.
In conclusion, the researchers have developed a broad framework for analyzing transcriptional dynamics that may be used in a wide range of biological systems. More broadly, dynamo can be combined with remarkable experimental advances in single-cell approaches, such as RNA metabolic labeling, lineage tracing, RNA age, signal pathway recording, and genetic perturbations. And in combination, it will facilitate to move forward into holistic kinetic theories and models of the entire organism for cell atlas initiatives, to understand how complex cell states arise from the combinatorial regulation of a limited number of factors, and finally to tackle the ultimate goal of converting between any cell types.
Story Source: Xiaojie Qiu, Yan Zhang, Jorge D. Martin-Rufino, Chen Weng, Shayan Hosseinzadeh, Dian Yang, Angela N. Pogson, Marco Y. Hein, Kyung Hoi (Joseph) Min, Li Wang, Emanuelle I. Grody, Matthew J. Shurtleff, Ruoshi Yuan, Song Xu, Yian Ma, Joseph M. Replogle, Eric S. Lander, Spyros Darmanis, Ivet Bahar, Vijay G. Sankaran, Jianhua Xing, Jonathan S. Weissman, Mapping transcriptomic vector fields of single cells, Cell, 2022.
Background Image Source: https://news.mit.edu/2022/new-computational-tool-predicts-cell-fates-genetic-perturbations-0203
Code Availability: https://doi.org/10.1016/j.cell.2021.12.045
Data Availability: https://github.com/aristoteleo/dynamo-release
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Swati Yadav is a consulting intern at CBIRT. She is currently pursuing her Master's from Delhi Technological University (DTU), Delhi. She is a technology enthusiast and has a keen interest in the latest research on bioinformatics and biotechnology. She is passionate about exploring new advancements in biosciences and their real-life application.





{kind=link}