
The rapid influx of large genomic data during the recent pandemic caused by COVID-19 constrained the researcher’s ability to analyze vast microbial genomes at once due to the lack of faster and more powerful tools. Researchers at EMBL-EBI have recently come forward with a robust bioinformatics tool MAPLE which efficiently reduces the size of bulky genomic data. The reduced size of the datasets helps to reduce dependency on computer memory and generate accurate phylogenetic analysis on sample data. This allows for the rapid analysis of vast genomic data all at once.
The recent COVID-19 pandemic highlighted the need to study genomic data obtained from pathogens to trace the transmission rate of such diseases successfully. It has also increased the significance of genomic surveillance of the fast-spreading COVID-19-causing SARS-CoV-2 variants and strains. The available genetic data in the case of the COVID-19 pandemic successfully helped not only in tracking the evolution, growth, and transmission of the SARS-CoV-2 virus but also efficiently identified mutations in the gene sequences of the virus that led to the origin of newer strains and variants.
A large amount of genetic data isolated from either the pathogen or host is required to understand the evolution and accurately predict the transmission of disease. The exploration of genomic data for epidemiological studies is largely dependent on the phylogenetic analysis of the organism.
Phylogenetic Inference to track genomic epidemiology
Traditionally, biologists used bioinformatics tools for inferring phylogenetic relationships to study the evolution of organisms from a specific lineage. The reconstructed phylogenetic trees were back-traced to track the evolutionary changes as well as the origin of a particular organism and identify the point of divergence of a subspecies. Evolutionary models are formed from phylogenetic trees.
The recent pandemic led scientists to utilize phylogenetic analysis to trace the evolution and transmission of the virus. The COVID-19-causing SARS-CoV-2 underwent rapid evolution over a short course of time, and keeping track of its evolutionary history as well as successfully identifying newer strains was rendered the most important part of epidemiological study. Rapid advances in phylogenetic analysis techniques have helped to determine whether the mutations in the variants have altered the biology of the virus. This data can guide researchers in formulating high-impact drugs and vaccines.
Maintaining updated phylogenetic trees with the rapid influx of sequence data was becoming increasingly challenging. Nextstrain, an open-source tool that stored pathogen genome data, failed to keep track of hundreds of genomic sequences generated daily during the global pandemic. With the limitations of existing tools, the requirement for new cutting-edge technology was needed to tackle the massive datasets generated during the pandemic.
The need for new cutting-edge technology
The surge of COVID-19 pandemic-related data exposed a major loophole in research, where most available bioinformatics tools and algorithms failed to analyze vast amounts of available data at once. The larger updated universal phylogenetic data are more accurate than local and smaller ones in predicting the disease outcome. Previously existing phylogenetic analysis programs like RAxML (Randomized Axelerated Maximum Likelihood) and the open-source software package IQ-TREE efficiently confer accurate phylogenetic inference for smaller datasets. Analyzing more extensive genomic data like that of the COVID-19 pandemic would take longer to generate predictions. To overcome the issues regarding the analysis of large-scale genomic data, scientists at EMBL-EBI suggested an algorithm called MAPLE which stands for ‘MAximum Parsimonious Likelihood Estimation.’
The MAPLE algorithm enables scientists to evaluate large-scale data all at once. According to the research paper published in Nature Genetics, the MAPLE algorithm works on organisms with closely related genomes. Maple performs phylogenetic inference by reconstructing evolutionary trees, which helps in analyzing the evolutionary history of the virus and can predict the transmission rate in a time-bound manner.
Phylogenetic trees are basically the diagrammatic representation of the evolutionary history and the evolutionary connection between species. The first step in performing phylogenetic inference is forming phylogenies of large sets of genomic data. This is achieved by performing multiple sequence alignments (MSAs) on the dataset. MSA is an algorithm to align sequences that are related to each other and share evolutionary relationships. MSA uses the FASTA format for their alignment files. The Data file for a sample in Fasta format includes the whole sequence of DNA, which increases the amount of data burden on computers. MAPLE utilizes a variant call format file (VCF file) in place of the FASTA format to minimize the size of the alignment file. Reduced size of input files requires low memory requirements and faster prediction of available genome datasets.
The MAPLE algorithm
The MAPLE algorithm performs phylogenetic analysis based on probability on the vast genomic data. MAPLE algorithm delivers rapid results along with reduced dependency on highly sophisticated supercomputers with vast memory. Having been extensively used to map genomic data from the recent COVID-19 pandemic, MAPLE has proved to be more precise in inferring results than the standard maximum likelihood algorithm. Analyzing extensive data with reduced dependency on computer memory was much faster.
In bioinformatics, the phylogenetic inference is one of the major problems faced by researchers as the available dataset is increasing day by day, thus posing a problem for researchers to develop algorithms to extract information from such a huge dataset. The recent COVID-19 pandemic exposed the limitations of standard phylogenetic methods (Bayesian and Maximum Likelihood methods) inability to scale large datasets. The MAPLE algorithm is applicable for sequences with slight deviations from one another in closely related genomes.
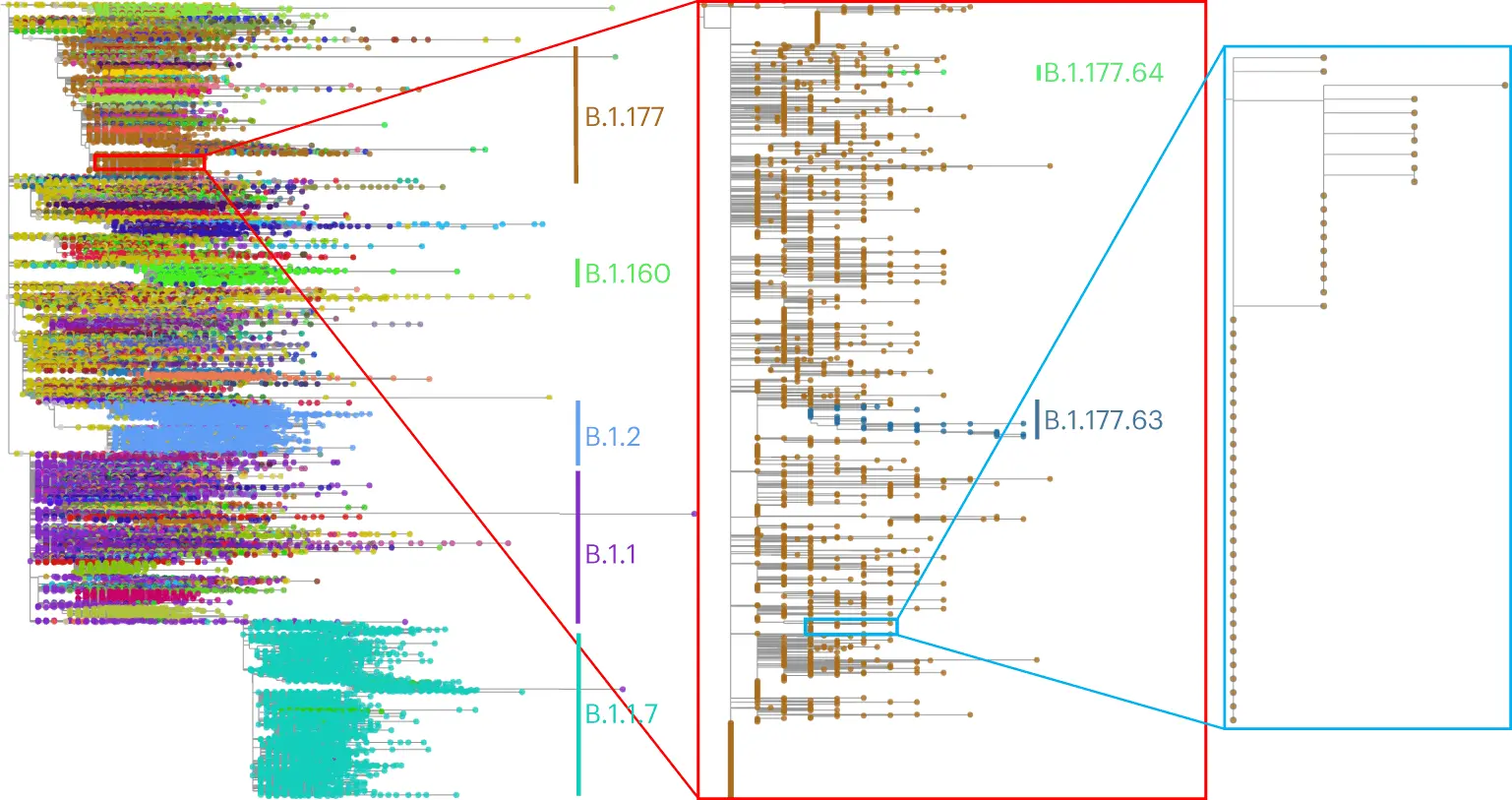
The following is an illustration depicting an estimate by MAPLE on sequence data from SARS-CoV-2

Image source: https://www.nature.com/articles/s41588-023-01368-0/figures/5
Conclusion
The MAPLE algorithm can reduce the size of bulky genomic data, thus constructing more concise phylogenetic inference in less time and reducing dependency on computer memory. Having successfully reduced the dependence on computer memory, MAPLE can devote its resources to accurately constructing phylogenetic trees in less time and with less memory than other standard approaches. This will enable the researchers to scale down large volumes of data once at a time.
Article Source: Reference Paper | Reference Article | MAPLE Availability: GitHub
Learn More:




Sipra Das is a consulting scientific content writing intern at CBIRT who specializes in the field of Proteomics-related content writing. With a passion for scientific writing, she has accumulated 8 years of experience in this domain. She holds a Master's degree in Bioinformatics and has completed an internship at the esteemed NIMHANS in Bangalore. She brings a unique combination of scientific expertise and writing prowess to her work, delivering high-quality content that is both informative and engaging.





{kind=link}