
Recent advancements in trajectory inferences have unfolded a new side of bioinformatics, i.e., single-cell analysis. The research group from the University of Northern Carolina School of Medicine benchmark study addressed gaps in capturing temporal changes in a single-cell state. Interpretation of dynamic phenotypes using ten datasets spanning different biological environments at three distinct temporal stages. The benchmark study predicted cell modalities and relative disease phenotypes.
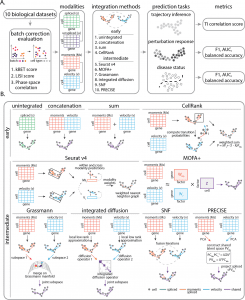
Image Source: https://doi.org/10.1186/s13059-022-02749-0
Trajectory inference methods have surfaced as a novel class of single–cell bioinformatics tools to investigate cellular dynamics. Transcriptomics information holds great promise in research due to its extensive data on individual cells to decipher their role in biological processes and disease etiology. Earlier research suggests single-cell analysis based on static states, which posed two fundamental challenges for understanding dynamic cell states. The first is an inability to distinguish between gene regulatory mechanisms due to similar mRNA measurements. Secondly, historical data often fails to capture the sizeable biological unevenness needed for population-level conclusions by missing other cell trajectories.
Ranek et al. presented the first task-oriented benchmarking study integrating ten datasets predicting cellular trajectories. Employing computational tool RNA velocity, which has been recently used to extract information from single-cell sequencing. The tool has been successfully incorporated into algorithms to deduce inferences mapping cell fate, gene regulatory networks coupled using scribe, and differentiation trajectories.
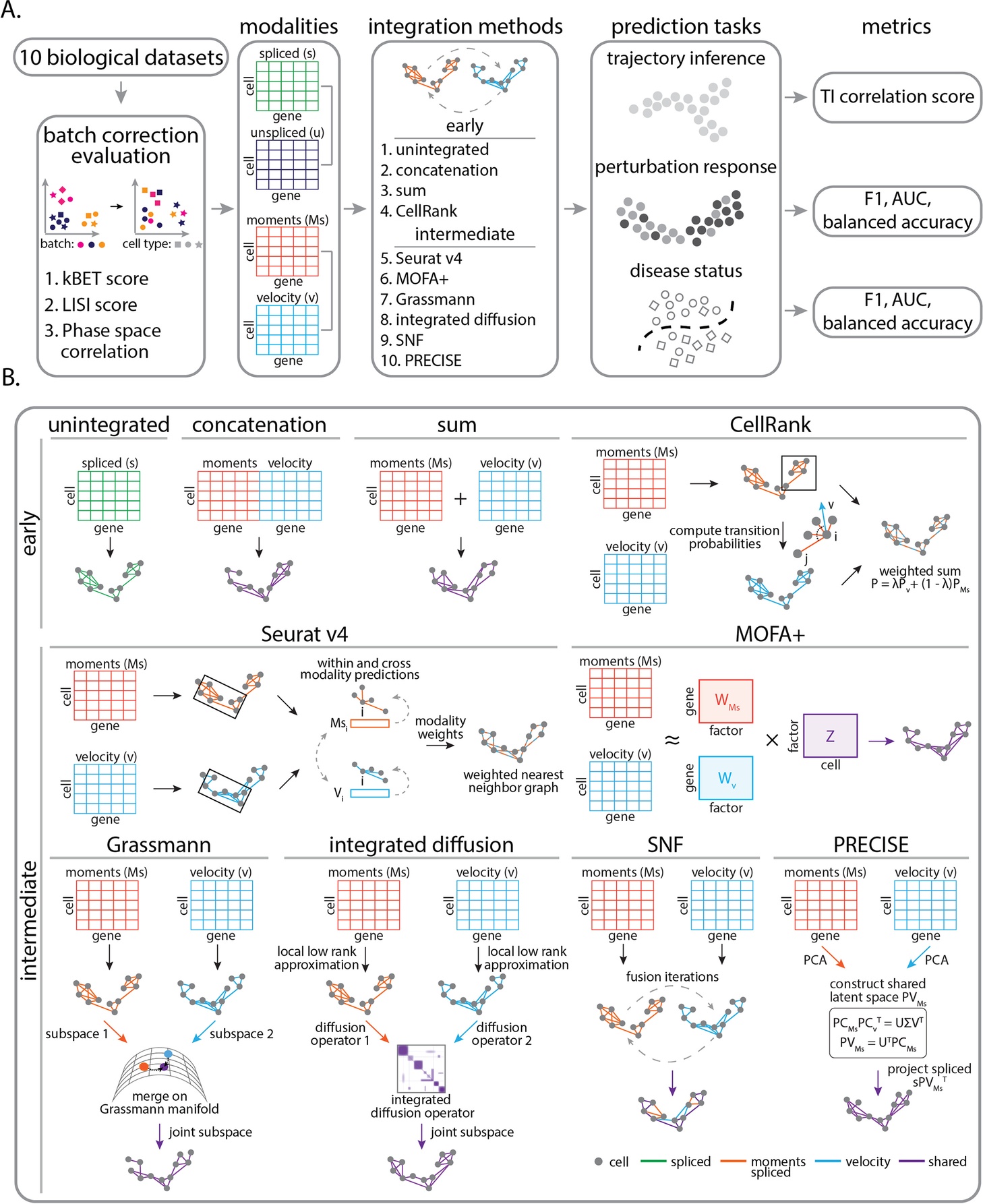
Overview of Benchmark Design
The team hence investigated the integration of gene expression modalities at the three distinct temporal stages of gene regulation: unspliced, spliced, and RNA velocity. Employing benchmark ten integration procedures on ten biological datasets with applications ranging from cellular differentiation to disease progression improves the predictive analysis.
The ten biological datasets included hematopoiesis differentiation, Mouse embryonic cell cycle, Natural killer T cell differentiation, LPS stimulation, INFγ stimulation, Acute myeloid leukemia chemotherapy, and diagnosis/relapse, Multiple sclerosis. Followed by integration approaches evaluation by selecting and grouping these approaches into two categories: early integration approaches and intermediate integration approaches. The early integration includes baseline bioinformatics strategies to merge individual possibilities into one input matrix.
The early integration methods included Unintegrated (A representation consisting of one data modality), Concatenation ( Modalities are merged through cell-wise linking of data matrices), Sum (Modalities are united through cell-wise summing data matrices), CellRank ( algorithm aims to model the cell state dynamics of a system). The intermediate integration approaches included Seurat v4 ( the R tool kit used for single-cell analysis), Multi-Omics Factor Analysis v2 (MOFA+)- integration of multi-omic data sets in to analyze variability, Similarity network fusion (SNF)- merges data by first computing a cell affinity graph for each data type, Grassmann joint embedding (to analyze structural datasets), Integrated diffusion (merges data modalities by first computing a diffusion operator for each denoised data type), Patient Response Estimation Corrected by Interpolation of Subspace Embeddings (PRECISE)- uses preclinical models.
As a result, of the two biological trajectories for cell cycle and differentiation, the researchers used gold standard cell type annotations for phenotypic markers. The trajectories for each integration approach were constructed by applying a partition-based graph abstraction (PAGA) topology-preserving map. The cell state transitions were also supported by the research literature upon inferring the predicted trajectories.
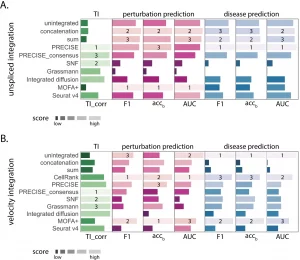
Image Source: https://doi.org/10.1186/s13059-022-02749-0
Upon evaluating integration for perturbation, considering single-cell RNA sequencing’s ability to identify subpopulations of cells relevant to drug therapy. Upon integration of unspliced or RNA velocity, the team tested performance in categorizing the perturbation from three diverse datasets with clinical application, which included the lipopolysaccharide (LPS) stimulated cells (macrophages), Interferon γ (INFγ ) stimulated pancreatic islet and peripheral blood mononuclear cells (PBMCs) from AML patient post-chemotherapy. The gene expression analysis of cell identification is associated with disease and recovery conditions.
The integrative analysis of the unspliced or RNA velocity data can help distinguish disease cell states by comparing diseased data against healthy individuals. The data sets included:
- Acute myeloid leukemia (AML).
- Multiple sclerosis (MS) case/control data.
- An MS case/control dataset of peripheral blood mononuclear cells (PBMCs).
As predicted and analyzed perturbation results, the unspliced integration achieved elevated accuracy for predicting the degree of disease.
Final Thoughts
The study sets the benchmark by providing trajectory inference as to how integrative approaches can be used using optimal parameters for the prediction of gene regulation expression. Both spliced and unspliced integration, including modalities, revealed cellular trajectories, e.g., cell division and differentiation. The features also supported the classification of cell subpopulations associated with disease states. The authors also acknowledged limitations that can be directed to future directions, such as a focus on evaluating the temporal gene expression integration to gain increased biological insight, such as unsupervised cell population identification and characterizing the phenotypic-related cells.
Article Source: Reference Paper
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Mahi Sharma is a consulting Content Writing Intern at the Centre of Bioinformatics Research and Technology (CBIRT). She is a postgraduate with a Master's in Immunology degree from Amity University, Noida. She has interned at CSIR-Institute of Microbial Technology, working on human and environmental microbiomes. She actively promotes research on microcosmos and science communication through her personal blog.





{kind=link}