

Emu, an approach that uses an expectation-maximization algorithm to generate taxonomic abundance profiles from full-length 16S rRNA reads profiles microbial populations using common genes.

Image Source: https://news.rice.edu/news/2022/emu-stands-tall-detecting-bacteria-species
According to results generated from simulated datasets and mock communities, Emu is capable of accurate microbial community profiling while obtaining fewer false positives and false negatives than alternative approaches,
When determining a microbe’s species, part of a gene is better than none. However, in their quest to develop a program that could identify every species in a microbiome, computer scientists from Rice University found that part was far from sufficient.
The microbial community profiling software Emu successfully identifies bacterial species by utilizing lengthy DNA sequences that cover the complete length of the gene being studied.
The Emu project led by the computer scientist Todd Treangen and the graduate student Kristen Curry of Rice’s George R. Brown School of Engineering makes it possible to analyze a crucial gene that microbiome researchers use to identify bacterial species that may be dangerous or beneficial to humans and the environment.
Their target, 16S, is a ribosomal ribonucleic acid (rRNA) gene component whose application was pioneered by Carl Woese in 1977. In bacteria and archaea, this region is highly conserved and contains variable regions critical for identifying distinct genera and species.
It is frequently employed for microbiome investigation because it is found in all bacteria and the majority of archaea, according to Curry, some areas have been preserved over time, making them simple to target. When sequencing DNA, we need some of it to be consistent across all bacteria so that we know what to search for, and then we need some of it to differ so that we can distinguish between various bacteria.
This study conducted by the Rice team with collaborators from German as well as the Houston Methodist Research Institute, Baylor College of Medicine, and Texas Children’s Hospital is published in the journal Nature Methods.
In the past, we tended to concentrate on harmful bacteria, or what we thought was nasty, and we didn’t really care about the others, says Curry. But in the last 20 years, there has been a change, and now we believe that some of the other microorganisms that are present may have some significance.
According to Curry, all the microscopic creatures in an environment are referred to as the microbiome. Commonly researched habitats include water, soil, and the intestines. Microbes have been found to have an impact on human health, carbon sequestration, and crops.
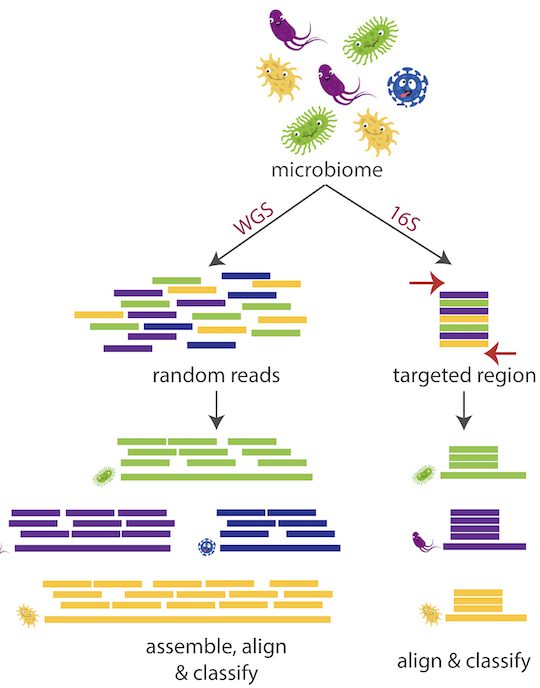
Image Source: https://news.rice.edu/news/2022/emu-stands-tall-detecting-bacteria-species
Emu, the name drawn from the function it performs “expectation-maximization,” analyses full-length 16S sequences from bacteria processed by an Oxford Nanopore MinION portable sequencer and uses sophisticated error correction to identify species based on nine different “hypervariable areas.”
With earlier technology, we could only read a portion of the 16S gene, said Curry. This gene comprises around 1,500 base pairs, with short-read sequencing, only 25 to 30 percent of the gene could be sequenced. To achieve species-level accuracy, however, the full-length gene is truly necessary.
But even the most cutting-edge technology has flaws, allowing errors to creep into sequences.
As stated by Treangen, an assistant professor of computer science who specializes in tracking infectious diseases, error rates have decreased recently, but they can still have up to 10% error inside an individual DNA sequence, while species can be differentiated by a few changes in their 16S gene. The critical computational challenge of this research effort was differentiating sequencing error from real variations.
One problem, the researchers noted, is that a lot of the error is nonrandom, which means it might repeatedly happen in particular places and start to appear as actual differences rather than sequencing errors.
A diverse variety of microbes can exist at abundances considerably below the sequencing error rate due to the thousands of bacterial species that might be present in a single sample, according to Treangen. This means that in order to separate signal from error, we can’t only rely on ad hoc cutoffs.
Instead, as Emu analyses microbial communities, it iteratively improves its error correction by comparing a large number of lengthy sequences, first against a template, then against one another. In comparative experiments, Emu dramatically reduced the number of false positives as compared to other methods when examining identical data sets.
According to Treangen, long-reads constitute a disruptive technology for microbiome research. The aim of Emu was to use the entire 16S full-length gene’s information without masking anything in order to see if the research team could make calls at the genus or species level that were more precise. And with Emu, we succeeded in doing just that, owing to a successful, cross-disciplinary joint effort.
Story Source: Curry, K.D., Wang, Q., Nute, M.G. et al. Emu: species-level microbial community profiling of full-length 16S rRNA Oxford Nanopore sequencing data. Nat Methods (2022). https://doi.org/10.1038/s41592-022-01520-4
https://news.rice.edu/news/2022/emu-stands-tall-detecting-bacteria-species
Learn More About Bioinformatics:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.











{kind=link}