
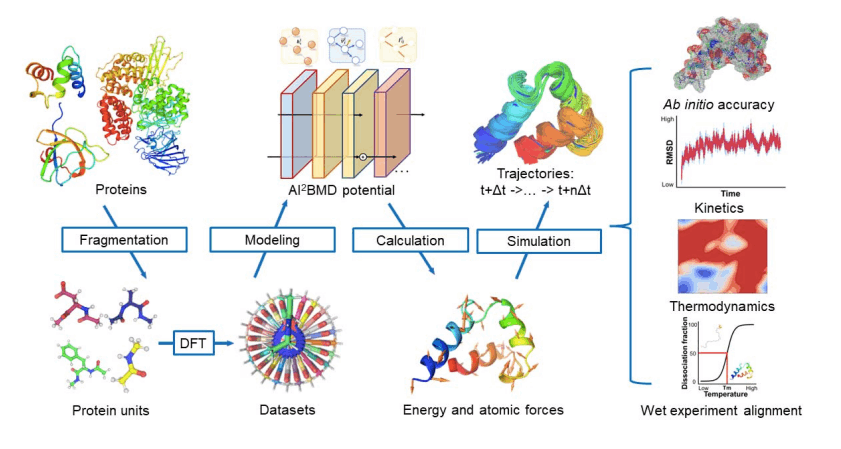
Microsoft Researchers from Beijing, China, have introduced an AI-Based ab initio Biomolecular Dynamics System (AI2BMD) for large biomolecule simulation by strategizing protein fragmentation scheme and machine learning force fields, which accomplishes a generalizable ab initio accuracy for energy and force calculations of proteins comprising over 10,000 atoms with a faster pace, reducing computational time. Being attributed with accurate predictions for potential energy and atomic forces for protein units, simulating conformational space exploration and protein kinetics, and capturing protein folding and unfolding tendencies, AI2BMD can anticipatedly complement lab experiments for inferring dynamism in the biomolecular realm.
A Glance into the Significance of Molecular Dynamics
The biological world’s essence lies in the dynamism of its molecules and their interactions. Understanding that dynamism is essential for every field of research. As Richard Feynman stated, “Everything that living things do can be understood in terms of the jiggling and wiggling of atoms,” but capturing those real-life movements is nearly impossible in analytical experiments.
Molecular Dynamics (MD), an extension of Molecular Mechanics (MM), blends laws of physics and simulations to circumvent the issue. In Molecular Dynamics, complex systems are computationally modeled at the atomic level by solving equations of motion numerically that accommodate the time evolution of the system, representing the kinetic and thermodynamic properties. MD is routinely operated for modeling the time-dependent motions or trajectories of biomolecules.
Background of the Study: Bringing Generalizable Solution to Molecular Dynamics Simulation of Different Proteins
In classical Molecular Dynamics, forces are calculated using a prescribed interatomic potential function, but one intrinsic constraint is its lack of chemical accuracy. Density Functional Theory (DFT), a quantum-mechanical method, calculates the electronic structure of atoms, molecules, and solids, which can attain chemical accuracy but is inapplicable to large molecular systems. In this regard, the authors draw attention to ab initio molecular dynamics (AIMD). In an AIMD calculation, finite-temperature dynamical trajectories are deduced using forces directly obtained from the potential derived from the electronic structure of the molecules.
AIMD brings feasibility to the study of chemical processes in condensed phases in an accurate and unbiased manner. AIMD encounters scalability and enormous time complexity issues. Machine learning-assisted methods were approached to solve the problem. However, the researchers pinpointed the deficiency in generalizability in those previous machine learning force fields (MLFF) approaches since the conformational space of a molecule is enormous. Therefore, training on limited conformations and adapting it for exploring conformational space was difficult to accommodate. Additionally, a lack of training data impedes the application of machine learning force fields for large biomolecules. In this context, AI2BMD (AI-Based ab initio Biomolecular Dynamics System) aspires to offer a unified generalizable solution for large molecule simulation, delivering ab initio accuracy.
An Overview of AI2BMD: Consolidation of Protein fragmentation scheme and Machine Learning Force Field
The framework first adopts a protein fragmentation approach, as tackling and generating the training data directly at the DFT level for whole large proteins is computationally prohibitive. Hence, the proteins are first fragmented into overlapping units to handle the complexity of deducing the associated energy and force of the conformation. Following the fragmentation technique, the intra- and inter-unit interactions are calculated and reassembled to specify the energy & force of the protein conformation. Thus, this fragmentation approach provides generalizability and feasibility in machine learning force field training.
Moreover, the researchers built a comprehensive dataset to train, test, and validate ViSNet (Vector-Scalar interactive graph neural Network) models that calculate the energy and atomic forces for the protein with ab initio accuracy. ViSNet was developed by the same research team in 2021, which is an equivariant geometry-enhanced graph neural network that extracts geometric features such as angles, dihedral torsion angles, and improper angles in accordance with the force field of classical MD with linear time complexity and efficiently models molecular structures with low computational costs.
Advantageous Features of AI2BMD
- In the assessments conducted by the researchers in terms of the comparative performance level of energy mean absolute error (MAE), AI2BMD has surpassed the conventional molecular mechanics (MM), AI2BMD has aligned more closely with DFT results, showcasing its capability in accurate predictions for both potential energy and atomic forces for protein units.
- AI2BMD has displayed a dramatic reduction in time complexity or computational time.
- Moreover, AI2BMD excels in deducing conformational space exploration and protein kinetics for both protein dipeptides and proteins, exhibiting its competency to bridge the discrepancy between simulations and experiments.
- It can capture conformational changes from a fully unfolded structure to a curled intermediate state and detects meaningful conformational changes and detailed interatomic interactions essential for the investigation of protein dynamics.
- It can execute free energy and melting temperature estimation of proteins, leading to reasonable predictions of protein folding thermodynamics.
Conclusion
Biomolecular simulation in ab initio accuracy is challenging yet holds the potential to derive insights into the trajectories of biological systems. In this scenario, the proposed framework attempts to eradicate the criticism of accuracy, robustness, and generalization ability with respect to the applicability of machine learning force fields. AI2BMD offers generalizability, adaptability, and versatility in simulations of different protein systems as it considers the fundamentals of proteins, i.e., stretches of amino acids, and fetches an improved energy/force calculation and kinetics/thermodynamic properties estimation.
Accordingly, AI2BMD can potentially assist in unraveling mechanisms of interactions, behavior, and activities of proteins as it can better snapshot flexibility in protein movements. The excellent performance of AI2BMD in evaluation studies has demonstrated its capability to complement wet-lab experiments. In contrast to classical MD simulation, AI2BMD lags in terms of efficiency. Future studies will try to ameliorate this constraint and extend AI2BMD in simulating lipids, nucleotides, nanomaterials, and solute-solvent interfaces for accentuating its utility in material science, drug discovery, and bioengineering.
Article Source: Reference Paper
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Aditi is a consulting scientific writing intern at CBIRT, specializing in explaining interdisciplinary and intricate topics. As a student pursuing an Integrated PG in Biotechnology, she is driven by a deep passion for experiencing multidisciplinary research fields. Aditi is particularly fond of the dynamism, potential, and integrative facets of her major. Through her articles, she aspires to decipher and articulate current studies and innovations in the Bioinformatics domain, aiming to captivate the minds and hearts of readers with her insightful perspectives.





 uses machine learning to characterize protein dynamics without experimental data.){kind=link}