
Machine learning-based methods have played a significant role in detecting RNA modifications. The advent of new high-throughput experimental and computational techniques has propelled the discovery of RNA modifications. The epitranscriptome refers to the biochemical modifications of the RNA (transcriptome) within a cell. Such modifications significantly impact several biological processes. The authors of the paper published in Briefings in Bioinformatics present a comprehensive review of machine learning methods for the detection of chemical messenger RNA modifications using diverse input data sources. The authors discuss strategies for training and testing these methods to encode epitranscriptome-relevant features as well as their interpretations. Challenges and open questions in the field of epitranscriptomics are also discussed, including the ambiguity in predicting RNA modifications in single nucleotides or transcript isoforms.
The What and How of Epitranscriptomics
Epitranscriptomics deals with studying RNA editing and modifications. The first documented evidence of an internal chemical modification of RNA was that of the discovery of pseudouridine as a fifth nucleotide in yeast RNA around sixty years ago. The technological advancement and cost reduction in high-throughput sequencing have greatly advanced our capacity to study these modifications in a transcriptome-wide manner. As of today, more than 150 internal RNA modifications have been discovered. Some of these have been found to be transcriptome-wide in protein-coding mRNAs. Some modifications are found to be reversible, meaning they can dynamically regulate RNA metabolism processes such as splicing, translation, export, and stability.
Of all modifications of RNA, m6A is one of the most abundant and well-characterized. These modifications show a strong enrichment around stop codons, and the deposition of m6A occurs mostly within DRACH sequence motifs. Methyltransferase writer complex deposits m6A on mRNA, and it is dynamically regulated, as is the binding of m6A by reader proteins as well as the removal by eraser enzymes. The combined effects of writers, readers, and erasers make m6A a key factor in the regulation of RNA processing steps, thereby impacting several physiological processes. However, the vast terrain of the epitranscriptome remains unexplored owing to the lack of methods for transcriptome-wide detection of these modifications, that are universal, fast, and highly reliable.
The development of transcriptome-wide experimental techniques for the detection of RNA modifications at the single-nucleotide level or the single-molecule level has shaped our understanding of the epitranscriptome. These methods use techniques such as targeted RNA modification detection using antibodies and enzymes or direct RNA sequencing to read RNA modifications in RNA molecules directly.
Computational methods developed for analyzing and interpreting these experimental results greatly involve the use of a machine learning-based approach. In the current article, the authors have outlined two major ML-based approaches for the detection of RNA modifications, viz., experiment-independent and experiment-based.
Experiment-Independent ML Methods for the Prediction of RNA Modifications
Experiment-independent methods do not require experimental data from high-throughput experiments that are designed to identify specific modifications. Such methods only require the reference sequence. In these methods, annotated RNA sequences and transcript characteristics are used as features for the prediction of modification tasks. The ML models are trained on already known, experimentally determined modification sites.
The most common paradigm used in such methods is that of SVM (Support Vector Machines), followed by the decision-tree-based algorithms RF (Random Forest) and XGBoost. More recent tools are implemented using the DL-based approach using CNNs, bidirectional gated recurrent units, and RNNs.
The following figure illustrates the workflow of experiment-independent tools.
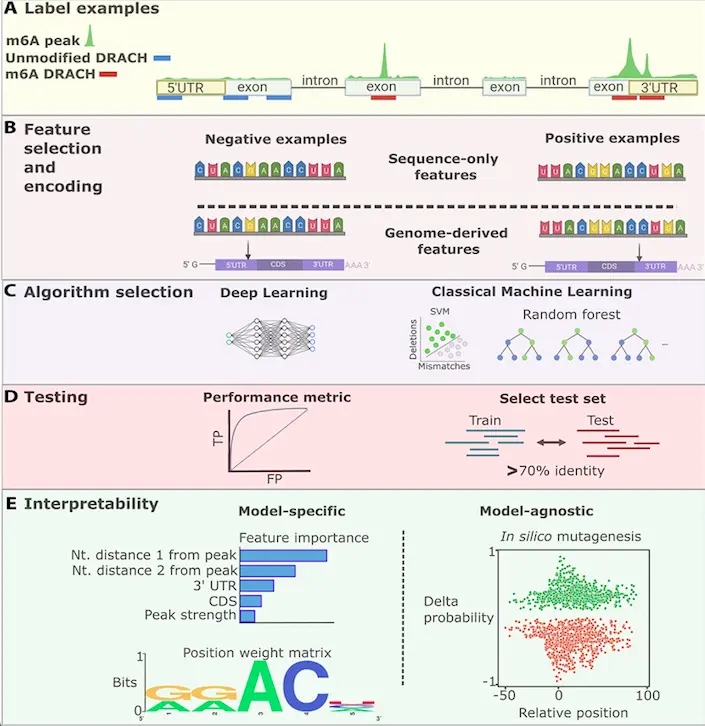
Image source: https://doi.org/10.1093/bib/bbad163
- These methods involve labeling RNA sequences to indicate the presence or absence of RNA modification based on experimental data.
- These methods can incorporate sequence-only features as well as a combination of sequence and RNA secondary structure.
- The selection of appropriate testing data and accuracy metrics for these methods is crucial for method performance analysis.
- The interpretability methods can be either model-specific or model-agnostic.
Such methods include WHISTLE, MethyRNA, and DeepM6ASeq, among others.
Experiment-based ML Methods for the Prediction of RNA Modifications
The authors use the m6A modification as an example to illustrate the experiment-based ML approaches for epitrancriptomics.
m6A detection from targeted experiments using ML approaches such as m6Aboost to obtain reliable m6A sites from experimental data. miCLIP2, like the original version, miCLIP was developed to detect m6A sites with single-nucleotide resolution using m6A-specific antibodies combined with targeted library preparation.
A classical ML-based method MAZTER-seq was developed for m6A site detection in an antibody-independent manner using RNase for cleaving.
DRS ( direct RNA sequencing)-based methods for detecting m6A modifications such as DRUMMER, ELIGOS, EpiNano, and JACUSA2 use the comparative approach for detecting the modifications. This involves translocating the RNA through a nanopore with the help of motor proteins.
Conclusion
The authors present a concise review of machine learning-based methods developed for epitranscriptomics. The experiment-independent methods, as well as the experiment-based methods, are both critical approaches, and their applicability is dependent on the research question at hand. While the experiment-independent methods are handy for investigating RNA modification sites similar to those in the training data as well as for a general characterization of the epitranscriptome, the experiment-based methods are better tailored for detecting RNA modifications under new conditions different from the training data. However, both approaches pose several challenges. The identification of isoform-specific RNA modifications is an open question in epitranscriptomics. Detection of RNA modification sites alone is not sufficient; rather, quantification in terms of stoichiometric measurements would provide a better and more precise picture of the epitranscriptome. RNA modifications across cell types remain elusive and offer opportunities for future investigations and method development. The need for high-quality experimental data for training the algorithms poses serious limitations. Nevertheless, machine learning has revolutionized our understanding of RNA modifications and, in the future, is expected to aid further understanding and uncovering of the epitranscriptome.
Article Source: Reference Paper
Learn More:




Banhita is a consulting scientific writing intern at CBIRT. She's a mathematician turned bioinformatician. She has gained valuable experience in this field of bioinformatics while working at esteemed institutions like KTH, Sweden, and NCBS, Bangalore. Banhita holds a Master's degree in Mathematics from the prestigious IIT Madras, as well as the University of Western Ontario in Canada. She's is deeply passionate about scientific writing, making her an invaluable asset to any research team.





{kind=link}