

Researchers from the University of Michigan, USA, developed a deep learning model, CAESAR (Chromosomal structure And EpigenomicS AnalyzeR), to predict nucleosome-resolution 3D chromatin contact maps from existing epigenomic features and lower-resolution Hi-C contact maps. The imputed high-resolution contact maps could help in target finding, hypotheses generation, and other downstream analyses.
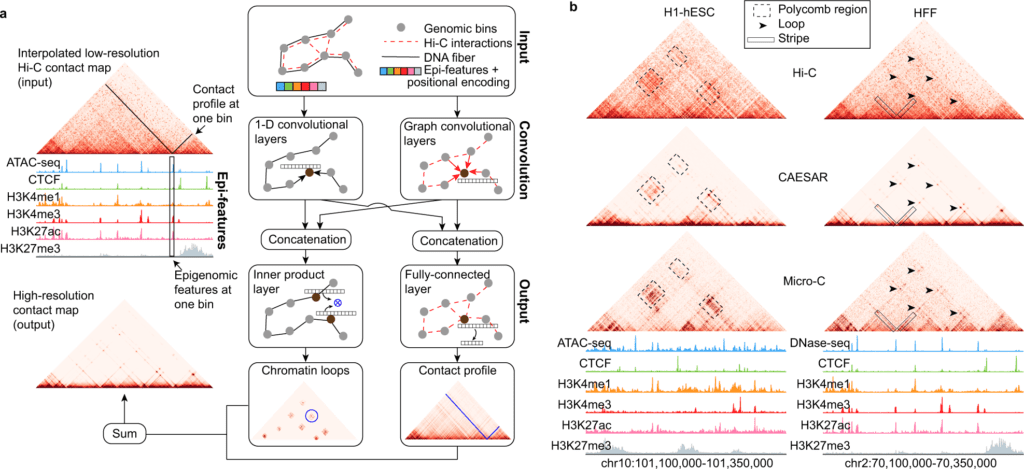
Image Source: Connecting high-resolution 3D chromatin organization with epigenomics
The resolution of chromatin conformation capture technologies continues to increase, and the new nucleosome resolution chromatin contact maps permit us to investigate how fine-scale 3D chromatin organization is connected with epigenomic states in human cells.
Utilizing openly accessible Micro-C datasets, the scientists created a deep learning model, CAESAR, to learn a mapping function from epigenomic elements to 3D chromatin organization.
The model precisely predicts fine-scale structures, for example, short-range chromatin loops and stripes, that Hi-C fails to detect. With existing epigenomic datasets from ENCODE and Roadmap Epigenomics Project, the scientists effectively imputed high-resolution 3D chromatin contact maps for 91 human tissues and cell lines.
In the imputed high-resolution contact maps, the researchers identified the spatial interactions among genes and their experimentally validated regulatory components, showing CAESAR’s actual capacity in coupling transcriptional regulation with 3D chromatin organization at high resolution.
Introduction to the CAESAR
While 3D chromatin organization at the large scale of topologically associating domains (TADs) and compartments has been well-characterized and described in numerous cell and tissue types by Hi-C technology, how the scientists interpreted fine-scale 3D chromatin organization at the nucleosome resolution had begun.
With the rising evidence that fine-scale chromatin organization at the nucleosome resolution is closely connected with the epigenomic state, one interesting question is whether we could precisely extrapolate such high-resolution chromatin contact maps from epigenomic features like chromatin accessibility, histone modifications, and transcription factor binding profiles. To investigate this, the scientists proposed the model CAESAR.
This model uses state-of-the-art deep learning methodologies to identify representations relevant to high-resolution chromatin organization. Specifically, 1D convolutional and graph convolutional layers distinguish epigenomic patterns over the linear chromatin fiber and over the 3D spatial chromatin organization that is pertinent to impute high-resolution chromatin contact maps.
With existing high-resolution Micro-C contact maps, Hi-C contact maps, and various cell-type matched epigenomic information on human H1-hESC (hESC), mouse ESC (mESC), and human foreskin fibroblasts (HFF), the scientists methodically assessed the model’s performance across various chromosomes, cell types, and species.
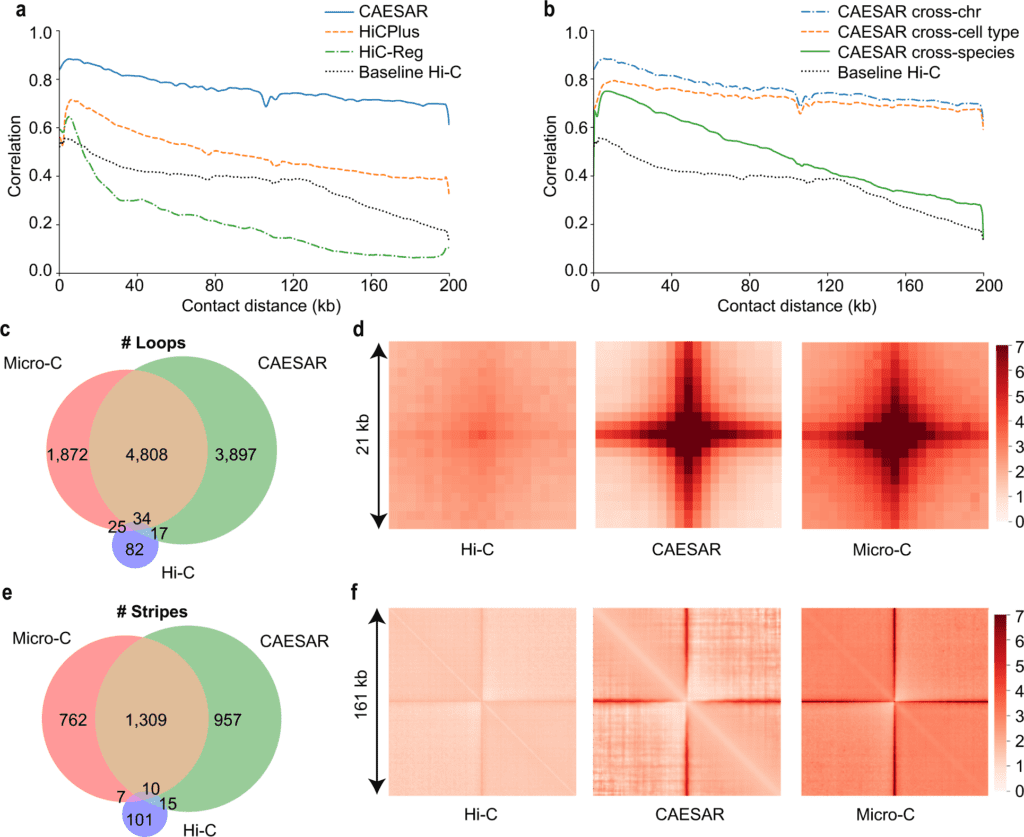
Image Source: Connecting high-resolution 3D chromatin organization with epigenomics
In the analyses, the model precisely imputes some fine-scale chromosomal structures that Hi-C sequencing fails to detect, including short-range chromatin loops and stripes. The model is more exact at imputing evolutionarily conserved regions, active A compartment, and early replicating regions, which indicates that the fine-scale 3D chromatin organization is firmly affected by the idea of the epigenomic factors in these regions.
The imputed chromatin contacts likewise recapitulate enhancer activities recently explained by CRISPRi experiments and manifest expression quantitative trait loci (eQTLs) previously profiled by the GTEx project.
CAESAR is additionally coupled with an attribution technique that distinguishes epigenomic features illustrative to these fine-scale 3D chromatin structures. The explanatory features help advance subtype fine-scale chromatin structures and elucidate the interplay between histone modifications and nucleosome level chromatin organization.
CAESAR connects 3D genome organization with epigenomics at nucleosome resolution and unprecedented scale. In the first place, compared with past computational models for imputing Hi-C contact maps, like HiCPlus10, HiCGAN11, and HiC-Reg12, CAESAR arrives at a much higher resolution.
Since most of the epigenomic activities (TF restricting and histone modifications) happen at the nucleosome resolution, creating a predictive model that associates epigenomics and chromatin organization at the nucleosome resolution is desirable.
Second, albeit past models EpiTensor13 and DeepTACT14 likewise reconstruct sparse 3D chromatin interactions from epigenomics at a super high resolution, CAESAR learns from genuinely Micro-C contact maps and predicts all chromatin contacts inside a distance range, which reveals diverse fine-scale structures like stripes, TADs, and polycomb interactions between repressive regions.
Third, unlike Akita15 and DeepC16, which predict chromatin contact maps from conserved DNA sequences, CAESAR produces tissue-specific or cell line-specific predictions from epigenomic features. Consequently, it imputes an uncommon number of high-resolution human chromatin contact maps, including 57 tissue samples, 16 cell lines, 12 primary cells, and 6 in vitro differentiated cells.
The imputed high-resolution contact maps are shared on a web server (https://nucleome.dcmb.med.umich.edu/), allowing clients to explore these fine-scale chromatin structures effectively and compare corresponding epigenomic features. Furthermore, CAESAR incorporates an attribution component, which uncovers detailed connections between 3D chromatin organization and epigenomic features.
The Takeaways from the Study
This research connects nucleosome-resolution chromatin structures with epigenomic features. Utilizing the presently accessible Micro-C contact maps for hESC, mESC, and HFF from the 4DN consortium and the corresponding epigenomic profiles from ENCODE and Roadmap Epigenomics Project, they systematically mapped 1D epigenomic profiles to fine-scale 3D chromatin structures with CAESAR.
The mapping was validated by high SCCs with observed Micro-C contact maps and the precise capture of fine-scale loops and stripes. CAESAR can be applied to produce high-resolution contact maps for any cell line or tissue as long as their common epigenomic features are profiled.
The model further connects transcriptomes with fine-scale structures and epigenomics by identifying the spatial interactions among genes and regulatory elements. In this manner, the imputed high-resolution contact maps will be helpful for target finding, hypothesis-generating, and other downstream analyses.
While CAESAR presents a strategy to examine fine details of 3D chromatin structure, the scientists noted that it is an advancing system with specific shortcomings which can be improved to the next level. To begin with, since Micro-C information, for the most part, outflanks Hi-C in the identification of short-range interactions, CAESAR likewise performs best at genomic distances of under 200 kb. Accordingly, CAESAR-imputed contact maps are not appropriate for investigations of large 3D chromatin structures like compartments.
Second, because Micro-C and Hi-C produce short-read sequences, the study is restricted to pairwise chromatin contacts, and accordingly, higher-order interactions are deficiently studied.
Third, their investigations showed that CAESAR performed well, as indicated by multiple evaluation metrics, yet there was a clear bias towards the A compartment, evolutionarily conserved regions, and early replicating regions. This is probably a reflection that the epigenomic features in the study are by and large more advanced in these regions. Thus, including additional epigenomic features might shift this bias impact accordingly.
Fourth, however, CAESAR demonstrated clear connections between epigenomic features and 3D fine-scale chromatin organization. The scientists didn’t notice a huge improvement in imputed contact maps with the rising number of epigenomic datasets. This proposes that epigenomic information may not make sense of the multitude of elements seen in 3D chromatin organization.
There might be unexplored layers of genetic and additional epigenetic data that assume a part in the organization of chromatin inside the nucleus. Up until this point, CAESAR demonstrated a system for the joint analysis of 3D chromatin structures and 1D epigenomic features at a matched resolution, and further integration of 1D DNA sequences is conceivable. For instance, the model might incorporate DNA sequences as features and elucidate 3D QTLs concerning high-resolution chromatin organization.
Paper Source: Feng, F., Yao, Y., Wang, X.Q.D. et al. Connecting high-resolution 3D chromatin organization with epigenomics. Nat Commun 13, 2054 (2022). https://doi.org/10.1038/s41467-022-29695-6
Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.











{kind=link}