
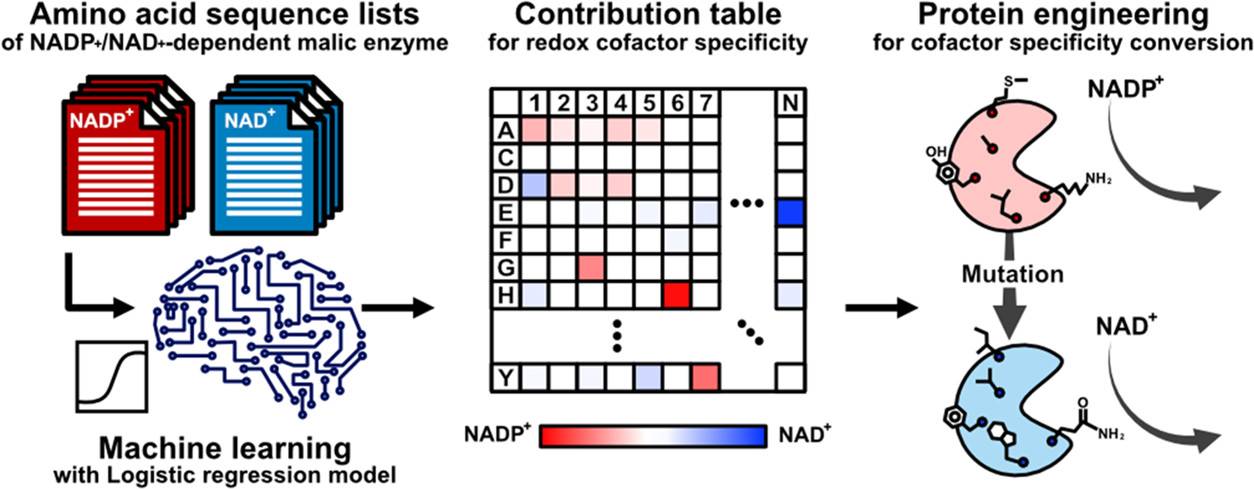
A recent study published in ACS Synthetic Biology has presented an analogous level of adaptability to enzymes. A team of researchers from Osaka University has illustrated the hypothesis to identify the amino acid residues actively involved in cofactor specificity by integrating a logistic regression model with an amino acid sequence dataset. The machine learning approach uses the redox cofactor conversion of the E. coli malic enzyme as a model that lacks an elucidated crystal structure allowing the model to develop a ranking index of amino acids that correlate to enzymes with distinct features.
Introduction
Proteins and constituting amino acids are both structurally and functionally important. The overall functioning and activity are based on their thermal stability, catalytic exertion, and substrate/cofactor specificity conversion depending on the environmental conditions. Enzymes execute extensive functions, enabled by the distinct arrangement of their constituent amino acids, but usually within a specific cellular environment. One of the long-standing research goals has been to modify or engineer enzymes artificially in order to allow the working of these enzymes in varied environments.
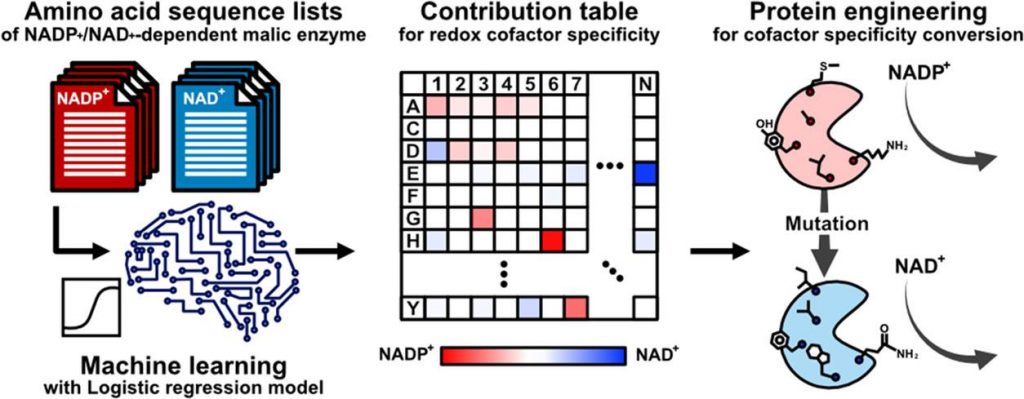
Image Source: https://doi.org/10.1021/acssynbio.2c00315
With computational biology approaches integrating the understanding using mathematical modeling and statistical learning the group of researchers from Osaka University Japan, have utilized the machine learning (ML) approach to assess enzyme function. Additionally, identify the location and number of amino acid residues that actively contribute to determining the substrate specificity distributed throughout the structure and not just the pocket of the substrate. The team hypothesized the possibility of conserved amino acid residues between the structurally analogous enzymes that also influences the different substrate or cofactor specificities.
The team has utilized the logistic regression model to develop a ranking index of the amino acids that correlate to enzymes with distinct features without having an illuminated crystal structure. The technological advancements of artificial intelligence and deep learning can significantly minimize the trial-and-error, but since it depends on experimentally obtained crystal structures of these enzymes that are often unavailable, the ML approach addresses it.
The researchers have tested the proposed hypothesis by focusing on the amino acids that are involved in the specificity of the Escherichia coli malic enzyme (ME) from the NADP+-dependent to the NAD+-dependent form for each position resulting in mutations. The results of the study have extensively highlighted the high accuracy and inferential approach of the machine learning model. The combined ML model approach with the phylogenetic analysis in order to determine the enzymatic design with the aim of adapting cofactor specificity.
The methodology was structured to understand the structural conservation of the malic enzyme (ME) to determine the structural similarity between the cofactor and different species. Various homologous folds were identified from different organisms for amino acid sequences of the ME obtained from the Protein Data Bank (PDB) which led to conclude that the functional conversion sequence exchange could be achieved if features of the NAD+ and NADP+-dependents could be extracted.
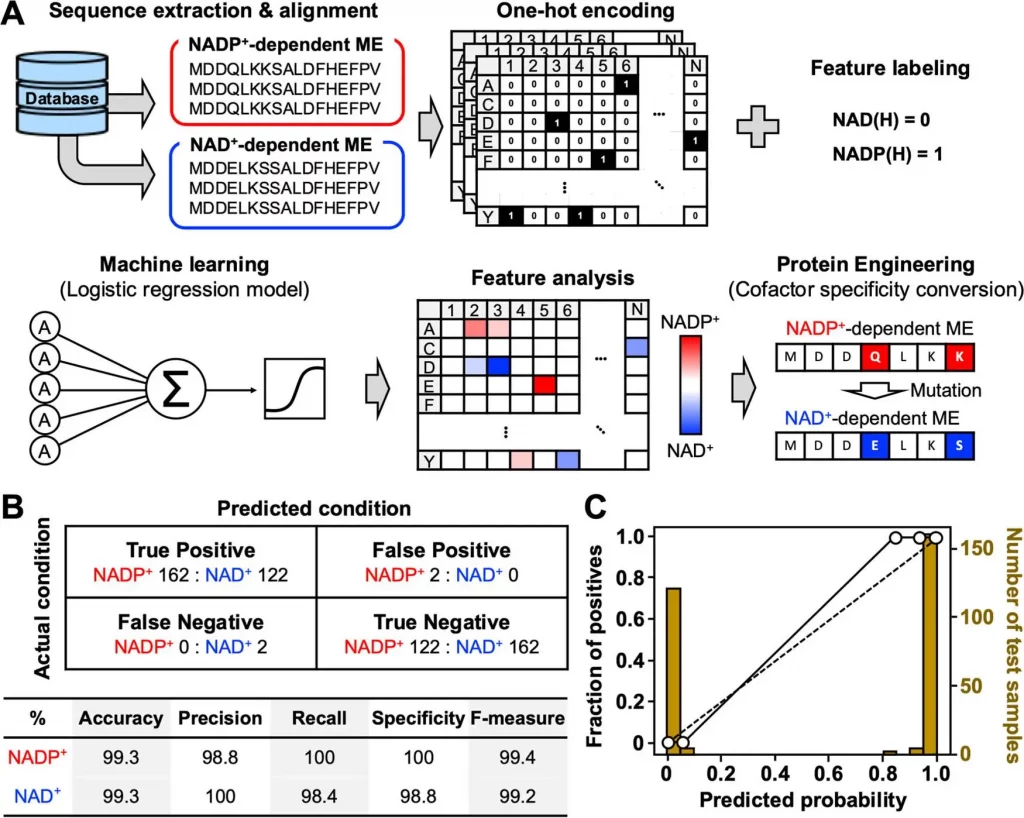
Image Source: https://doi.org/10.1021/acssynbio.2c00315
In order to construct the machine learning model, the team randomly selected sequences of both NADP+- and NAD+- dependents of E.coli enzyme MEs from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. The investigation aimed to determine the mutation position of the candidate amino acids for cofactor preference conversion. Approximately One thousand ME amino acid sequences were obtained, and 952 (448 NAD+-dependent and 504 NADP+-dependent) eccentric sequences were collected by ignoring the duplicates. The amino acid sequences within the dataset ranged around 289 and 1042 residues in length while unique sequences were aligned to detect the mutations such as point mutation, insertion, and deletion using Clustal Omega.
Post logistic regression model training, the partial regression coefficient (β) is a weight parameter for the cofactor specificity of each amino acid residue and is identical to the contribution of each amino acid residue to cofactor specificity. The research team has constructed a study on the cofactor specificity of ME from E. coli strain K12 (MaeB) considering the 3D structure for (MaeB) has not been elucidated.
In order to assess the catalytic property of the designed ME variants, the ML model introduced approximately 100 mutations into the trcMaeB to create mutants. The ML-designed trcMaeB mutants were prepared using an E. coli expression system, and only trcMaeB10-70 were soluble and obtained in a purified state. The ML model successfully displayed that 20−30 mutations were sufficient to switch the cofactor specificity of trcMaeB, and the expression level was not greatly affected. The model helped in narrowing down both mutation position and amino acid candidates that could not be deduced alone using the structural information.
In the molecular modeling simulations for structural, comparison, and visualization of the mutation sites, the comparative models were generated using Rosetta to identify the position of mutation and the effect of mutagenesis on the overall structure. The results suggested a tendency for hydrophobic amino acids to have appeared surrounding the substrate pocket. In trcMaeB10, R302G and D304A mutations were also observed. While substitutions around the pocket also display a slight change in internal structure and dynamics, which affects cofactor selectivity considering the mutations to change the hydrophilic amino acids to hydrophobic amino acids.
Future Perspectives
The Research work conducted at Osaka University has extensively succeeded in utilizing both AI and ML to hasten and improve progress by significantly reconfiguring an enzyme’s specific action mechanism without considerably altering the enzyme’s function. Future advancements in enzyme engineering will majorly benefit biomedicine and oil fields such as pharmaceutical and biofuel production. Since these require careful tuning of the versatility of enzymes to fit various biochemical environments.
Story Sources: Reference Paper
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Mahi Sharma is a consulting Content Writing Intern at the Centre of Bioinformatics Research and Technology (CBIRT). She is a postgraduate with a Master's in Immunology degree from Amity University, Noida. She has interned at CSIR-Institute of Microbial Technology, working on human and environmental microbiomes. She actively promotes research on microcosmos and science communication through her personal blog.





{kind=link}