
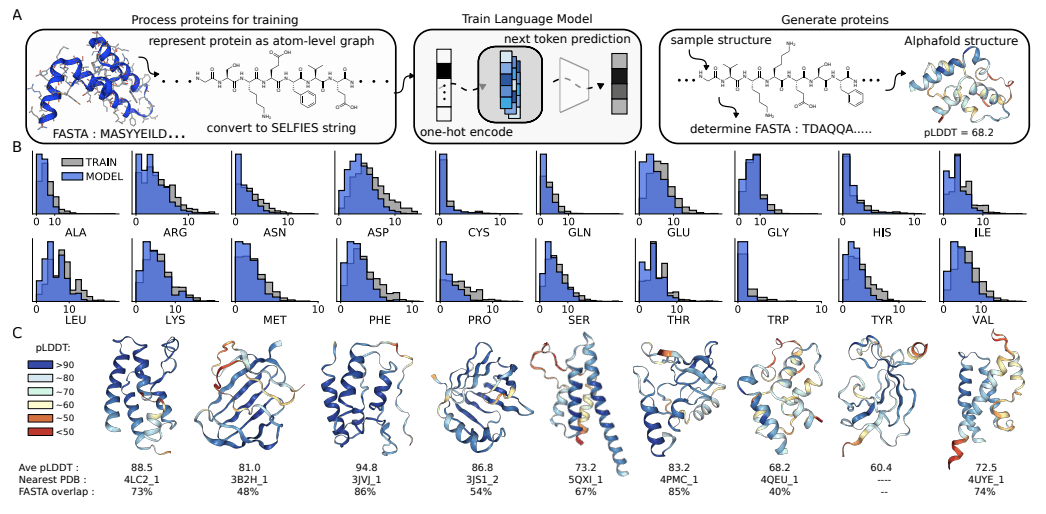
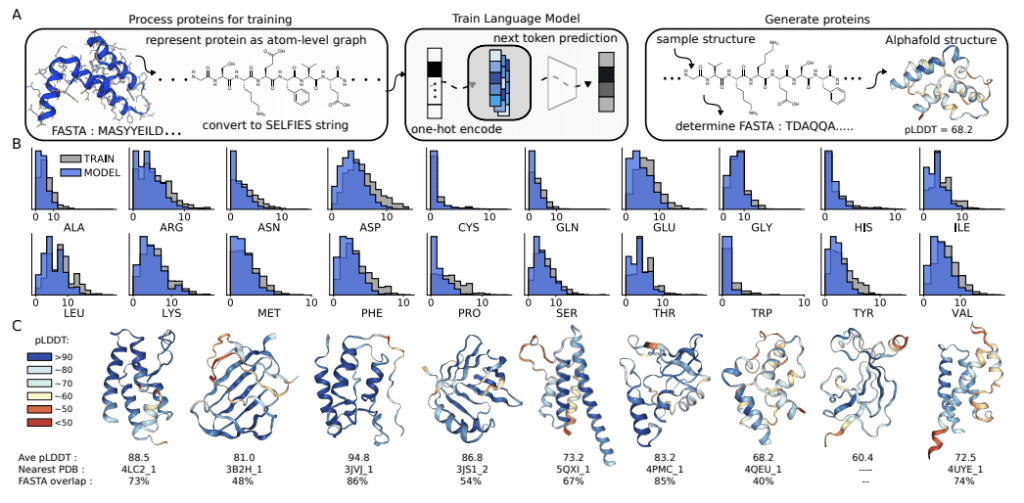
The University of Toronto researchers have explored the potential of Chemical Language Models (CLMs) to thrive as Biological Learning Models. In contrast to popular Protein Language Models that learn from protein sequences, CLMs can learn atomic-level representations of proteins and also learn protein backbones and natural amino acid structures, as well as the primary sequence patterns in the training sets. The study demonstrates CLMs’ capability to generate proteins having unnatural amino acids and novel protein-drug conjugates and thereby displays the prospects of expanding the domain of biomolecular design altogether and augmenting the representations of the combinatorial space of biology and chemistry.
The Relevance of the Emphasis on Protein Structure Prediction
Proteins are the workforce of the cell, and the entire cellular machinery is essentially protein-dependent. The sophisticated functionality of the protein is bestowed by the three-dimensional structural conformation of the protein. Envisaging protein structures empowers scientists to comprehend the mechanisms of cellular processes. Accordingly, perturbations in protein expression directly affect the cell’s physiology and ensues disease conditions. For these reasons, proteins are the most conceivable and capitalized targets for docking drug molecules and initiating drug-induced favorable modifications, also because modulation of nucleic acids through drugs in order to achieve desired results is rarely feasible.
Exploration of all these vibrant areas of life sciences starts with the same conventional requirement, which is to get acquainted with the structures of proteins. After the protein is isolated following an elaborate procedure, the next step is to understand the protein’s sequence and structure. The wet lab experimental approaches like X-ray crystallography, NMR, and cryo-EM have assisted in these tasks for decades. With the advent of computer-aided technologies, numerous attempts have been made to decrypt protein’s structural figures. Paving breakthroughs in Artificial Intelligence (AI) technology advancements, Language Models are now one of the greatest trending and leading providers and propellers of modeling proteins’ structures in silico.
Protein Structure Modeling Leveraging Language Models
Scientists have already cracked the enigma of protein’s infrastructure. Twenty definite types of Amino acids chained in different sequences are the fundamentals of each protein. Considering each amino acid as an alphabet, the relative orders of these amino acid alphabets govern the subsequent folding of the amino acid stretches into three-dimensional motifs and domains, which are the functional blocks of a protein, akin to words, phrases, and sentences in human language. This forms the very essence of Protein Language Models (PLM), which elaborates on the analogy between proteins and human languages, typically comprising modular elements that can be reused and rearranged.
These PLMs are developed by feeding protein sequences and experimental structures from protein databases. Therefore, PLMs recognize the pattern, relationships, and interdependencies between amino acid stretches that correspond to higher-order confirmation like helices, sheets, domains, and motifs of the protein molecule. Accordingly, as enunciated by the researchers from the University of Toronto, the PLMs neglect the atomic level interactions of the amino acids. Atoms being the finer substructure, protein representations at the atom level would expand the province of protein design. In this context, the researchers illustrate the applicability and pertinence of Chemical Language Models (CLM) in learning the language of proteins.
Highlight of the Study: Chemical Language Models in Generating Protein Models
CLMs are deep neural networks trained by masking or next token prediction, which use atom-level linear strings of sequences parsed from molecular graphs. These sequences represent entirely a molecule by including all atoms, bonds, rings, aromaticity, branching, and stereochemistry. These models can now generate larger complexes apart from small drug-like molecules, although these complexes are much smaller as compared to proteins. This recently revealed capability of CLMs to learn complex molecular distributions has been extrapolated by the researchers to make an attempt at atom-level representation of proteins.
CLM can learn the language of proteins entirely from scratch by learning to generate atom-level sequences that define proteins with valid primary sequences corresponding to meaningful secondary and tertiary structures. The authors demonstrate that CLM can generate proteins atom by atom with valid secondary and tertiary structures. More interestingly, further studies with proteins that have random sidechain modifications, creating proteins with unnatural amino acids, show that the model learns the continuous atom-level properties of the training proteins, including the octanol-water partition coefficient, the exact molecular weight (MW), the topological polar surface area (PSA), and the number of carbon, nitrogen, and oxygen. Additionally, the model learns a similar sidechain structure to the training sidechains. Moreover, it can generate proteins attached to small molecules, for instance, antibody-drug conjugates.
Conclusion
CLMs can learn multiple hierarchical layers of molecular information, including atom-level molecular properties, residue-level constraints for backbone and amino acid structure, primary sequence patterns, and motifs, and are capable of generating protein structures as sequence representations of atom-level graphs that are similar to the training proteins in the PDB (Protein Data Bank). CMLs can explore chemical and biological space together and simultaneously, as evidenced by their capability of modeling proteins with unnatural amino acid sidechains and protein-small molecule conjugates, respectively. The researchers anticipate that further studies should focus on the proteins with longer sequences and consider the 3D structure of the biomolecules while employing CLMs.
Article Source: Reference Paper
Important Note: arXiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Aditi is a consulting scientific writing intern at CBIRT, specializing in explaining interdisciplinary and intricate topics. As a student pursuing an Integrated PG in Biotechnology, she is driven by a deep passion for experiencing multidisciplinary research fields. Aditi is particularly fond of the dynamism, potential, and integrative facets of her major. Through her articles, she aspires to decipher and articulate current studies and innovations in the Bioinformatics domain, aiming to captivate the minds and hearts of readers with her insightful perspectives.











{kind=link}