
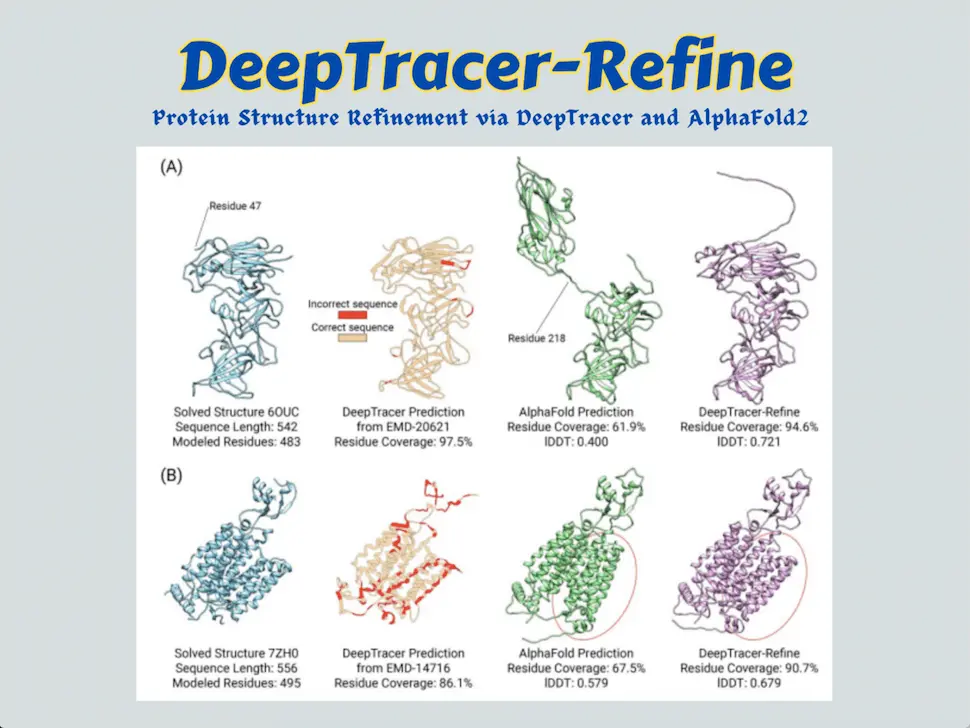
Scientists of the DeepTracer project have formulated DeepTracer-refine, an automated pipeline that consolidates the advantages of sequence-to-model and map-to-model strategies, complements their shortcomings, and aspires to refine Alphafold2-generated structures through amelioration of the regions with lesser confidence by splitting AlphaFold’s structure into compact domains and manually docking them into experimental maps to fix the residue locations.
A Glimpse into Protein Folding: Protein’s Three-Dimensional Structure
Proteins are the ultimate decryption of the genomic codes and workforces of a cell. The advent of sequencing techniques and spectroscopic methods hasn’t left any mystery about the constituents of proteins. Fundamentally, proteins are stretches of amino acids stringed together through peptide bonds.
More interestingly, all proteins are different combinations of only 20 amino acids. Regardless of the similarity in the components, proteins perform diverse, sophisticated functions. Therefore, to understand protein’s roles in biological machinery, it is important to acknowledge the essentiality of three-dimensional structures that make a protein functional.
Hence, generating protein sequences alone can’t help us understand a protein’s importance, knowing its native form or how the protein is actually there inside a cell is extremely important and intriguing for any field of life science. Here comes the phenomenon of protein folding.
When the ribosome complex translates the gene transcript, the proteins are synthesized by adding amino acids against a codon. Surprisingly, the non-covalently bonded interactions like hydrogen bonding, hydrophobic interactions, ionic bonding, van der Waal’s interaction, etc, between amino acids render the specialized 3D structure. Even the secondary structures, fundamental to protein’s 3D structures, share similarities. Now all the proteins are represented by the orientation of their secondary structures, which are the alpha helix and beta sheets.
Protein folding can be a self-assembly phenomenon, or correct protein folding can be mediated by other proteins, such as chaperons, present inside cells, exclusively dedicated to this work. The correct conformation of a protein is also governed by thermodynamic feasibility with respect to the respective cellular microenvironment. Proper folding makes the protein useful for executing its normal function. Visualizing a protein in its 3D form is fascinating and absolutely necessary for understanding a protein, and it makes gathering comprehensive knowledge about the protein’s structure possible.
Protein Structure Acquisition: Different Approaches
X-Ray crystallography and Nuclear Magnetic Resonance techniques have been employed by structural biologists for decades to capture protein’s 3D structure because proteins are too small to be visualized under typical microscopes. The technique that has had a profound impact on structural biology in this century is Cryo-EM or Cryogenic Electron Microscopy. It involves flash freezing of protein samples followed by generating microscopic images of each molecule by employing electrons, enabling near-atomic resolution.
The revolution in resolution has boosted the procurement of protein’s sophisticated structures. The advancement of computational tools has attempted to supplement the wet lab-based protein structure determination process. Considering the constraints of pursuing experimental methods, computer-assisted prediction of structures has empowered biologists to acquire insights about a protein without stepping into the laboratory. Deep Neural Network-based AlphaFold2, created by DeepMind, attained the best performance in the 14th critical assessment of structural prediction (CASP) round and is now an integral part of every researcher in the life sciences domain. The impact of AlphaFold2 is splendid.
The referenced paper enunciates two kinds of structural modeling strategies: Map-to-Model and Sequence-to-Model. From density maps produced in Cryo-EM techniques, the structure is decoded, and this can be termed as the former mentioned Map-to-Model method, where computer-aided tools automate the protein structure modeling from cryo-EM density maps in a template-dependent or independent manner. AlphaFold2 utilizes the latter approach, in which structures are modeled from protein sequences, introducing evolutionary information and energy functions.
Map-to-Model Method and Previous DeepTracer Model: Facility and Constraints
In the Map-to-Model method, recognizing the amino residues followed by sequentially connecting the residues to obtain the backbone is challenging. DeepTracer leverages a convolutional neural network called the U-Net to identify residues from cryo-EM density maps and employs a traveling salesman algorithm to trace the backbone, which connects the closest residues based on Euclidean coordinates. Sometimes the cryo-EM map doesn’t cover the entire protein, or there might be missing residues that limit the platform’s performance.
Also, experimental maps are noisy in nature and vary in size and quality. DeepTracer has attempted to overcome the issue by pre-processing the density maps into consistent input, but the problem of overfitting arises. On high-resolution and high-quality density maps, DeepTracer can achieve above 90% residue coverage, however, it is the missing 10% that creates challenges for DeepTracer. Moreover, DeepTracer performs a sequence alignment between the predicted and true sequences to eliminate any possibility of false connections and incorrect type predictions resulting in an inaccurately predicted sequence. But if the quality of the backbone trace is perturbed, it influences the accuracy of the model.
Implications of DeepTracer-Refine Pipeline: Consolidation of AlphaFold2 and DeepTracer
Although the Sequence-to-Model methods don’t suffer from lacking residue coverage, the particular drawback of ALphaFold2 is to inability to fold the regions in between domains accurately. Regarding the intrinsic pros and cons of these two strategies, DeepTracer-refine aspires to incorporate the advantage of perfect sequence coverage of AlphaFold2-generated structures and improve the less accurate backbone regions by utilizing DeepTracer’s prediction.
AlphaFold generates a per-residue metric called the predicted Local Distance Difference Test (pLDDT), from which the regions modeled with less confidence can be estimated.
The automated pipeline, DeepTracer-Refine, detects optimal locations to split the AlphaFold2 structure into compact domains by harnessing the pLDDT value and making iterative alignments to improve the predicted structure.
The refinement process follows an iterative split-align-merge procedure, in which every possible detected splitting location is attempted, and the last round of the iterative split-align-merge process is the final DeepTracer-Refine output. Moreover, it prevents false alignments like overlapping alignments, in which the compact domains are aligned to the same location, and distant alignments, in which two domains are aligned so far apart that they cannot be physically connected.
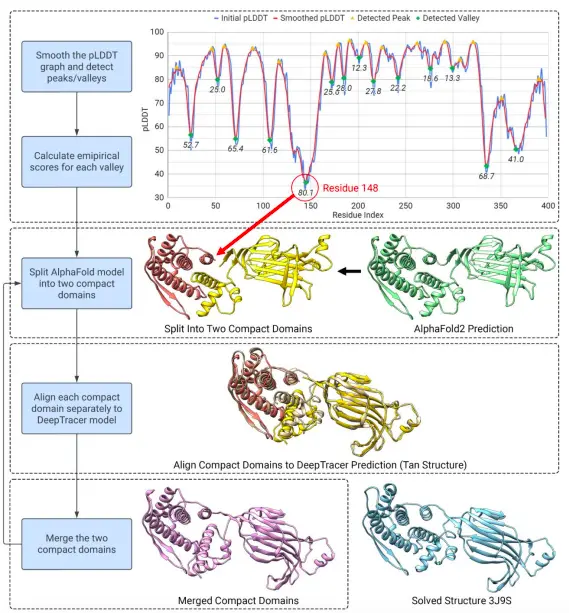
Image Source: https://doi.org/10.1101/2023.08.16.553616
Conclusion
DeepTracer-Refine increased the overall average residue coverage from 77.8% to 90.0% and the average lDDT from 0.667 to 0.707 from AlphaFold2’s initial prediction. Out of the 39 targets, it improved 27 structures according to the increase in residue coverage and lDDT. Upon comparison against Phenix’s AlphaFold refinement, this method exceeds Phenix in run-time performance. Some of DeepTracer’s predictions contain faults, for instance, it cannot fix the issues if there are imprecise folds within a protein domain itself or imprecise individual residues on the backbone. Nevertheless, DeepTracer-Refine extraordinarily conjugates the two prevalent strategies of in-silico protein modeling, complements their advantages, and thus offers a novel proposal for overcoming their respective flaws.
Article Source: Reference Paper
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Aditi is a consulting scientific writing intern at CBIRT, specializing in explaining interdisciplinary and intricate topics. As a student pursuing an Integrated PG in Biotechnology, she is driven by a deep passion for experiencing multidisciplinary research fields. Aditi is particularly fond of the dynamism, potential, and integrative facets of her major. Through her articles, she aspires to decipher and articulate current studies and innovations in the Bioinformatics domain, aiming to captivate the minds and hearts of readers with her insightful perspectives.











{kind=link}