

Scientists from the Wellcome Sanger Institute, Hinxton, UK, and the University of Tartu, Estonia, have developed a machine learning-based algorithm, MinsePIE, to predict prime editing insertion efficiencies. The authors performed a comprehensive analysis of prime editing efficiencies by designing a library of 3604 sequences of varying lengths and measuring the frequency of insertion into four genomic sites. The authors discovered a complex relationship between insertion sequence features and efficiency, shaped by DNA repair and processing mechanisms. The ML-based algorithm incorporates these findings to predict insertion efficiencies accurately and thus provides a catalog of insertion rates for several useful sequences. In the rapidly evolving field of prime editing, MinsePIE holds promise in facilitating both genome engineering and therapeutics.
Prime editing: the new entrant in the genome editing toolbox
Prime editing involves the insertion of short DNA sequences that do not require an external template and do not generate double-strand breaks. Prime editors consist of a nicking version of Cas-9 fused to a reverse transcriptase domain, which is complexed with a prime editing guide RNA, pegRNA. The pegRNA consists of a primer binding site homologous to the sequence in the target as well as a reverse transcriptase template, including the intended edit. While CRISPR-Cas9 is analogously comparable to molecular scissors, prime editors can be thought of as analogous to word processors capable of performing search and replace operations on the genome directly.
The prime editing process involves several partly independent steps, including three DNA binding events and successful DNA repair. These events are greatly influenced by the introduced sequence, which adds to the complexity of the editing system. The determinants of the efficiency of the editing system are also not fully understood. In the largest study so far performed to determine these determinants, the authors of the study comprehensively tested the influence of varying the reverse transcription templates as well as primer binding site lengths. The extensive study incorporated around 55,000 pegRNAs. GC content, melting temperature of primer binding sites, and Cas9 guide RNA activity were found to be determinants of the efficiency of the prime editing system. However, the majority of the libraries used in the study had the same single nucleotide substitution. Other studies undertaken to determine the efficacy of prime editing have largely focused on single nucleotide substitutions. Consequently, little is known about the effects of the inserted sequence on the efficiency of the prime editing system.
The authors conducted an extensive study involving experimental techniques to measure the insertion efficiency of 3604 sequences in several target sites under a variety of cellular and repair pathways contexts. Insertion sequence length, secondary structure, and nucleotide compositions are all found to be determinants of the insertion efficiency of the prime editing system. Variations in insertion rates were found to be best explained by sequence features and repair pathway activity. With these insights, the authors developed and trained an ML-based algorithm based on sequences to predict editing outcomes for novel sequences accurately. This aids in the selection of optimal reagents for new insertions.
Insights on the determinants of efficiency
The authors systematically characterized the effects of the length and composition of the insert sequence, cell line, target site, and version of the prime editor system on the insertion rates.
The insertion efficiency was calculated as defined by the authors, ”the fraction of reads in the target site amplicon with given insertion divided by the fraction of reads for the pegRNA encoding it in the pegRNA amplicon,” the main statistic in this study.
Insertion efficiencies of sequences were found to vary widely.
- Insertion frequency did not decrease monotonically with insert length in the HEK293T cells. The longest sequence that was inserted with >1% frequency was 66 nt, implying that it is feasible to insert moderately long sequences with prime editing.
- The mismatch repair (MMR) proficiency is found to be the prime source of independent variation between different cellular contexts for prime editing.
- The authors found that the overexpression of the 3’flap nucleases TREX1 and TREX2 inhibits the insertion of long sequences.
- The secondary structure and Cytosine content of the inserted sequence were both found to be positively correlated with the insertion rate.
MinsePIE: Modeling insertion efficiency for prime insertion experiments
The MinsePIE model was trained with XGBoost, which is an implementation of the gradient-boosted trees algorithm, one of the widely used machine learning techniques currently. The training involved the following features: length, MMR proficiency, percentages of nucleotides C, A, and T, the normalized secondary structure of the reverse transcriptase template, and many more such features. For training the model, unique sequences were divided into groups of training and testing sequences at a ratio of 0.7. Information based on measurements from different target sites and cell lines was also incorporated.
The following figure illustrates the steps in determining the efficiency of the prime editing system.
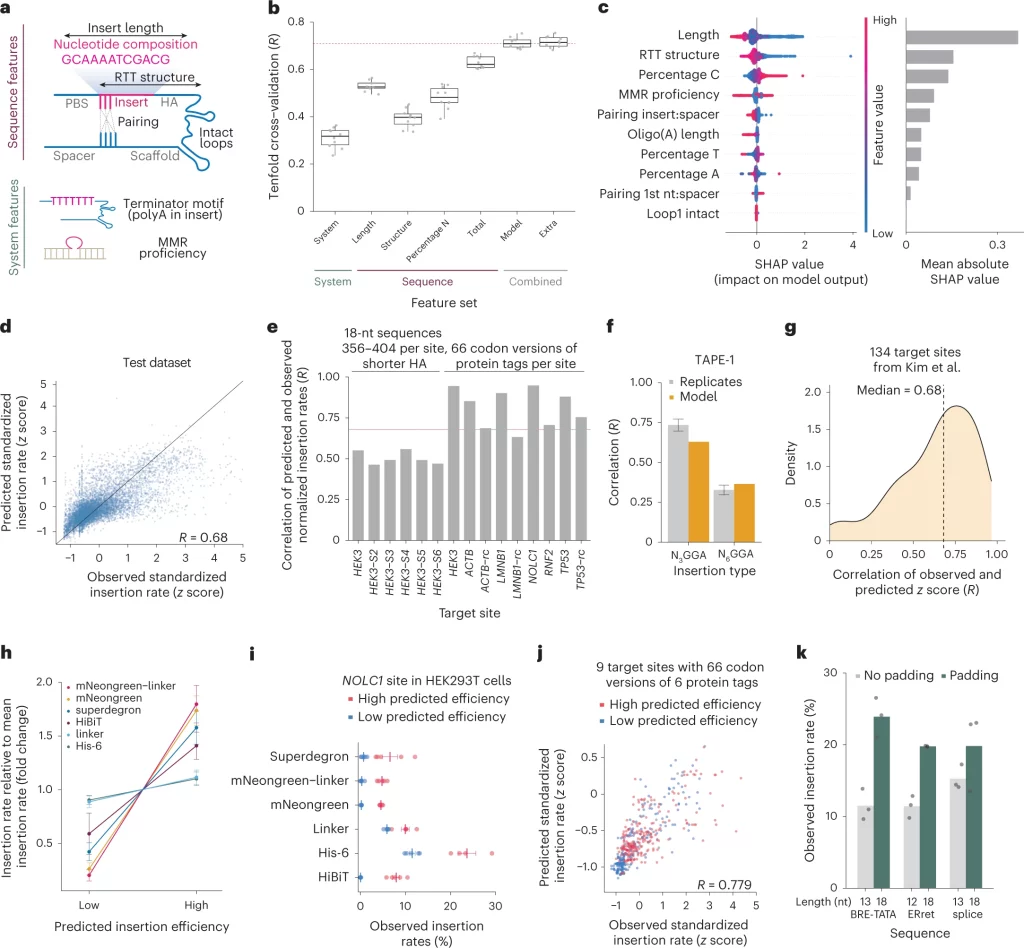
Image source: https://www.nature.com/articles/s41587-023-01678-y/figures/5
Conclusion
Short sequences are frequently and precisely inserted into a specific genomic target site. However, the determinants of the efficiency of insertion remained elusive until the authors developed the algorithm, MinsePIE. The authors perform a comprehensive analysis of 3604 pegRNA sequences to gain insights into the factors governing the insertion efficiency of the prime editing system. With the insights from the diverse experiments performed, the authors incorporated the sequence and repair features into a machine-learning algorithm to predict insertion efficiencies for novel sequences. While the method makes it feasible to apply prime editing to many more useful sequences, however, it is not devoid of limitations. The authors did not assay genome-wide off-target editing or model the insertion of nontemplated or mutated sequences. Nevertheless, MinsePIE is shown to predict insertion efficiencies accurately and provides a thorough understanding of the determinants of the prime editing system. The algorithm promises to add a new dimension to genome engineering and therapeutics.
Article Source: Reference Paper | MinsePIE: Website
Learn More:




Banhita is a consulting scientific writing intern at CBIRT. She's a mathematician turned bioinformatician. She has gained valuable experience in this field of bioinformatics while working at esteemed institutions like KTH, Sweden, and NCBS, Bangalore. Banhita holds a Master's degree in Mathematics from the prestigious IIT Madras, as well as the University of Western Ontario in Canada. She's is deeply passionate about scientific writing, making her an invaluable asset to any research team.





{kind=link}