
The introduction of NGS transformed the field of biology, allowing for greater insights into biological mechanisms through the gathering of rich, accessible data. However, important context is lost due to the data being conventionally stored in tabular formats, which erase the impacts that spatial connectivity has on genes and gene expression. A new framework, GENIUS (GEnome traNsformatIon and spatial representation of mUltiomicS data), developed by the Aarhus University researchers, shows that the incorporation of this crucial information may lead to improved performance in neural network models.
The invention of next-generation sequencing has entirely transformed the field of genetics through a combination of lowered costs and extensive data generation. NGS techniques are often performed repeatedly on samples in order to derive data about different aspects of the biological architecture, which can then be integrated to produce holistic conclusions about the samples being studied.
Current Obstacles in the Use of NGS Data
It is known that the data within the genome is organized in a spatial manner, positioned sequentially on the length of the chromosomes. However, genomic data derived through NGS is stored in a tabular manner instead, resulting in the loss of the spatial connectivity that occurs naturally in organisms.
Additionally, though the data generated is rich, the samples used are generally far smaller than the available feature space. When the number of such features that need to be evaluated grows, so does the chance association risk. In order to correct for the errors that occur in multiple hypothesis tests, p-value adjustments are applied such that the majority of results obtained are discarded, only allowing the most significant of the lot to remain. This is already considered a significant obstacle in the analysis of single-omics analysis but is drastically exacerbated in multi-omics data analysis. Commonly, multi-omics analysis involves the processing of observations on an individual basis and, in doing so, neglects to take into account the wealth of contextual information that can be gained through the consideration of how spatial organization and connectivity affect the genome.
Potential Solutions & Drawbacks
Artificial intelligence techniques have recently been spotlighted as potential fixes to the problem: Their introduction into medical applications has been significantly slower due to the abundance of regulatory mechanisms compared to other fields, such as finance or advertising. Despite this, artificial intelligence has been understood as a powerful tool to aid in bioinformatics research. Specifically, deep neural networks have shown great promise due to their ability to replicate non-linear data patterns and trends without needing additional correction when used in multiple-hypothesis testing. In addition, it has been observed that convolutional layers, when present in networks, significantly impact performance by reducing the influence of noise. Despite this, the common theme underlying their use is that such models struggle with interpretability. While simpler models may have better interpretability, they are also not capable of accurately depicting the non-linear patterns present within the data. Hence, black box models are often used as an alternative despite their low interpretability. However, interpretability is of high significance in the medical field, as the discovery of biomarkers and the identification of complex biological mechanisms is still an ongoing task.
GENIUS: The Creation of A Novel Framework
While there has been much progress in advancing the interpretability of these models in the form of novel frameworks like DeepExplain and DeepLIFT, there wasn’t yet a framework for the purpose of conducting multi-omics analysis such that patterns could be found in data that was feature-rich, multi-layered, and connected spatially. Using such connections resulted in vastly improved performance. Additionally, when used in combination with the Integrated Gradients framework, the contribution of features relative to each other could be evaluated in order to reach a more complete understanding of the intricate biological mechanisms that allow for classification by deep learning models.
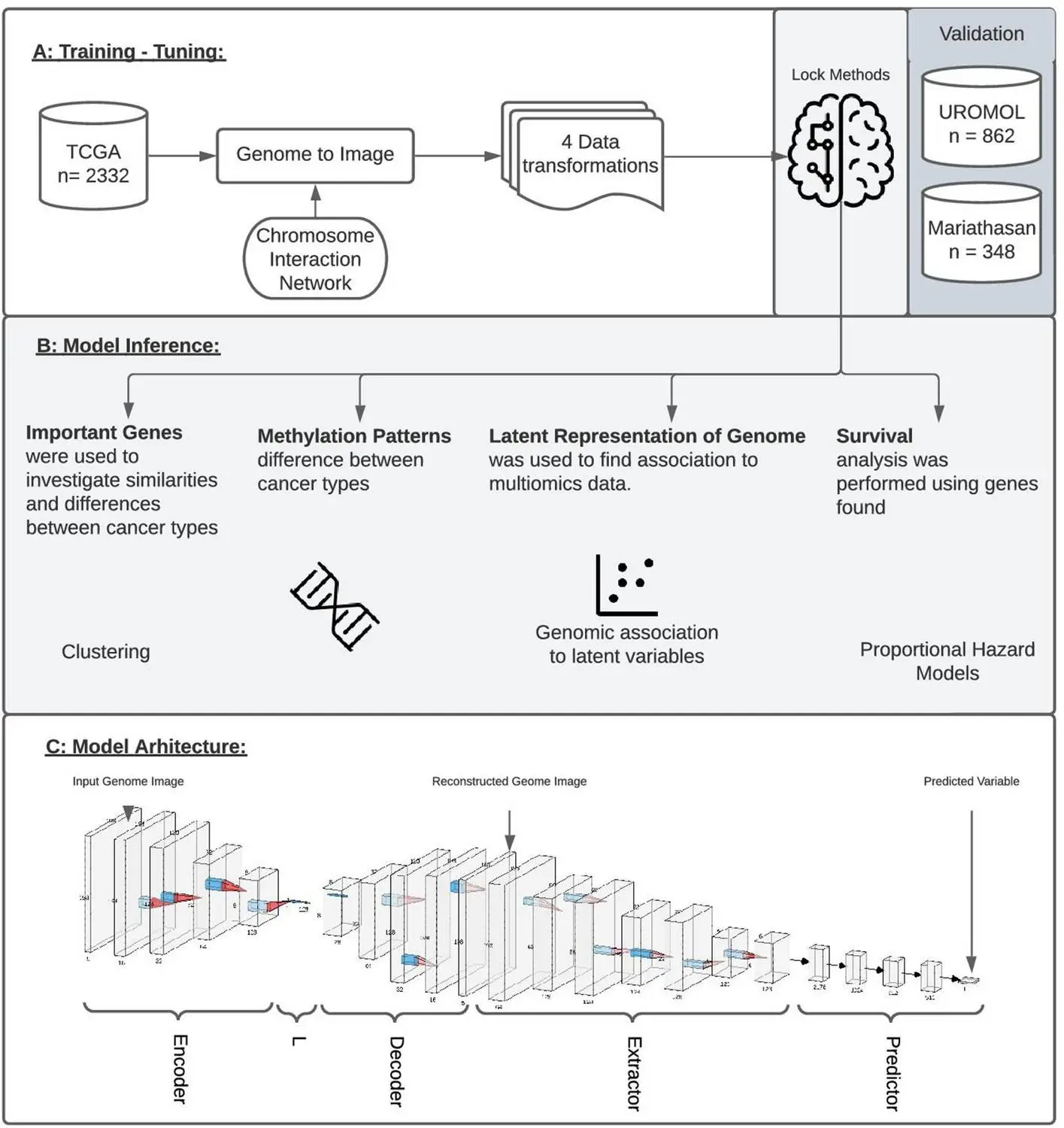
Using Integrated Gradients allows for the performance of a single analysis to produce results instead of a series of multiple tests. Data is transformed into genome images, the model is trained accordingly, and it is then inspected using IG to produce an attribution score for all the genes in the image. These can then be ranked relative to each other such that a set of genes that are most associated with respect to the output is recovered. This new framework is named GENIUS, and it has a two-part methodology: firstly, classification is performed, where the data is transformed into an image such that a single gene is depicted as a pixel within a genome “image,” after which multiple -omics data types can be incorporated as separate layers within the image. This is then used to train the deep learning model. Then, attribution scores are assigned, which allows the user to extract data and information about the features that influence a particular prediction made by the model.
The model was then tested on data available in Cancer Genome Atlas (TCGA): six models were created using GENIUS to predict metastatic cancer, the presence of the p53 oncogenic mutation, the tissue of origin, patient age, the presence of the chromosome instability marker wGII, and randomized tissue of origin. Through this, several different factors that affected the model’s predictions could be identified, which agreed with results from prior studies (for instance, the top 10 genes identified as the top drivers for metastasis of tumors in certain kinds of cancer have been previously shown to have a high impact on patient outcomes). Output layers and loss functions were changed as necessary in order to predict different variables. It was found that four of the six models were able to successfully use genome images as input in order to make accurate predictions, except in the case of age and randomized tissue of origin. Different image layouts were also tested for all six models, with all achieving similar results, though further research may be required in order to find an optimal organization method.
Conclusion
The provision of spatial information to deep learning networks is observed to vastly increase the accuracy and performance of neural network models. When tested on data available in TCGA, the network was able to identify and extract different factors that influence the development of cancer in patients, as well as specific genes whose presence determines patient response to immunotherapy, the tendency for metastasis, and overall disease outcome. This has important implications for the medical field, where the use of vast amounts of feature-rich data to identify patterns and trends has become increasingly common. Computational methods such as this may also significantly cut down costs associated with analysis and research.
Article Source: Reference Paper | All code is available on the public GitHub repository
Learn More:




Sonal Keni is a consulting scientific writing intern at CBIRT. She is pursuing a BTech in Biotechnology from the Manipal Institute of Technology. Her academic journey has been driven by a profound fascination for the intricate world of biology, and she is particularly drawn to computational biology and oncology. She also enjoys reading and painting in her free time.











{kind=link}