
Scientists from the University of Michigan employed hypergraph theory, utilizing long sequence reads to map genome-wide multi-ways to identify chromatin architecture within the human genome. Understanding how a cell is organized could help biologists develop treatments that halt cancer and other diseases.
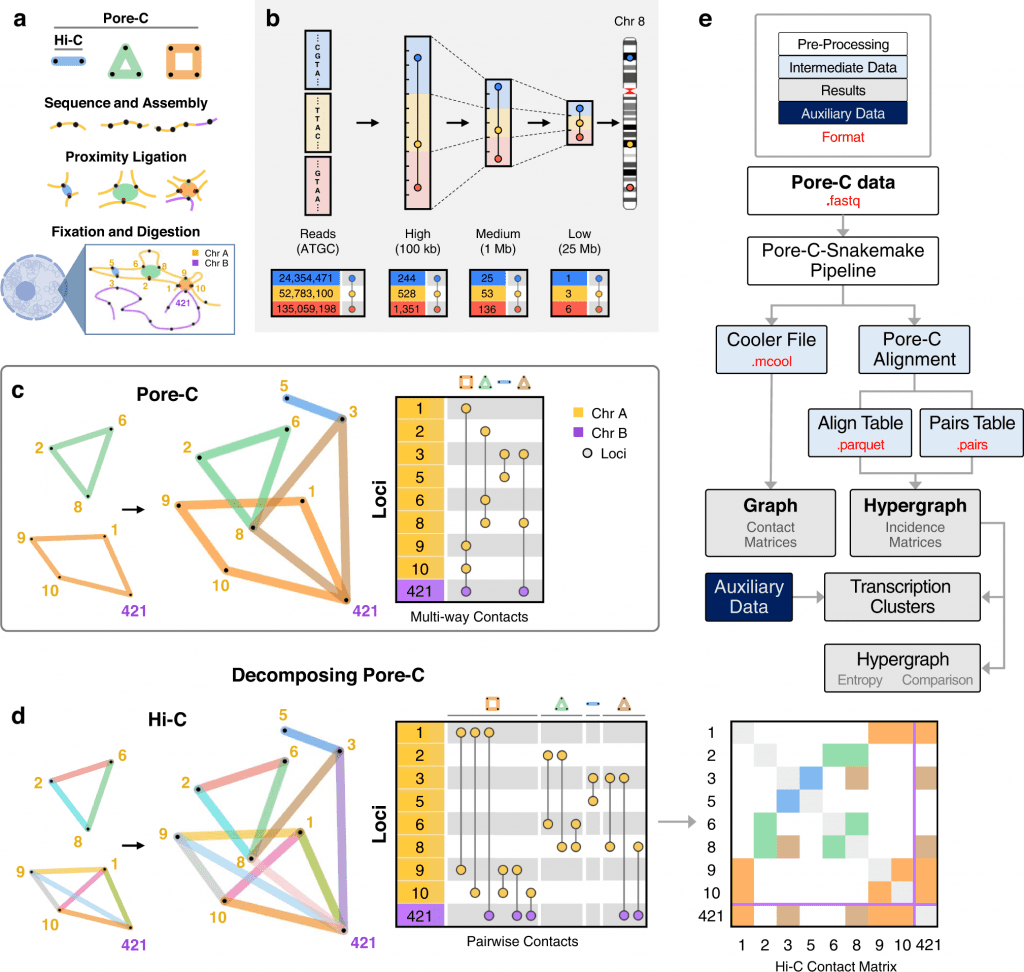
Image source: https://doi.org/10.1038/s41467-022-32980-z
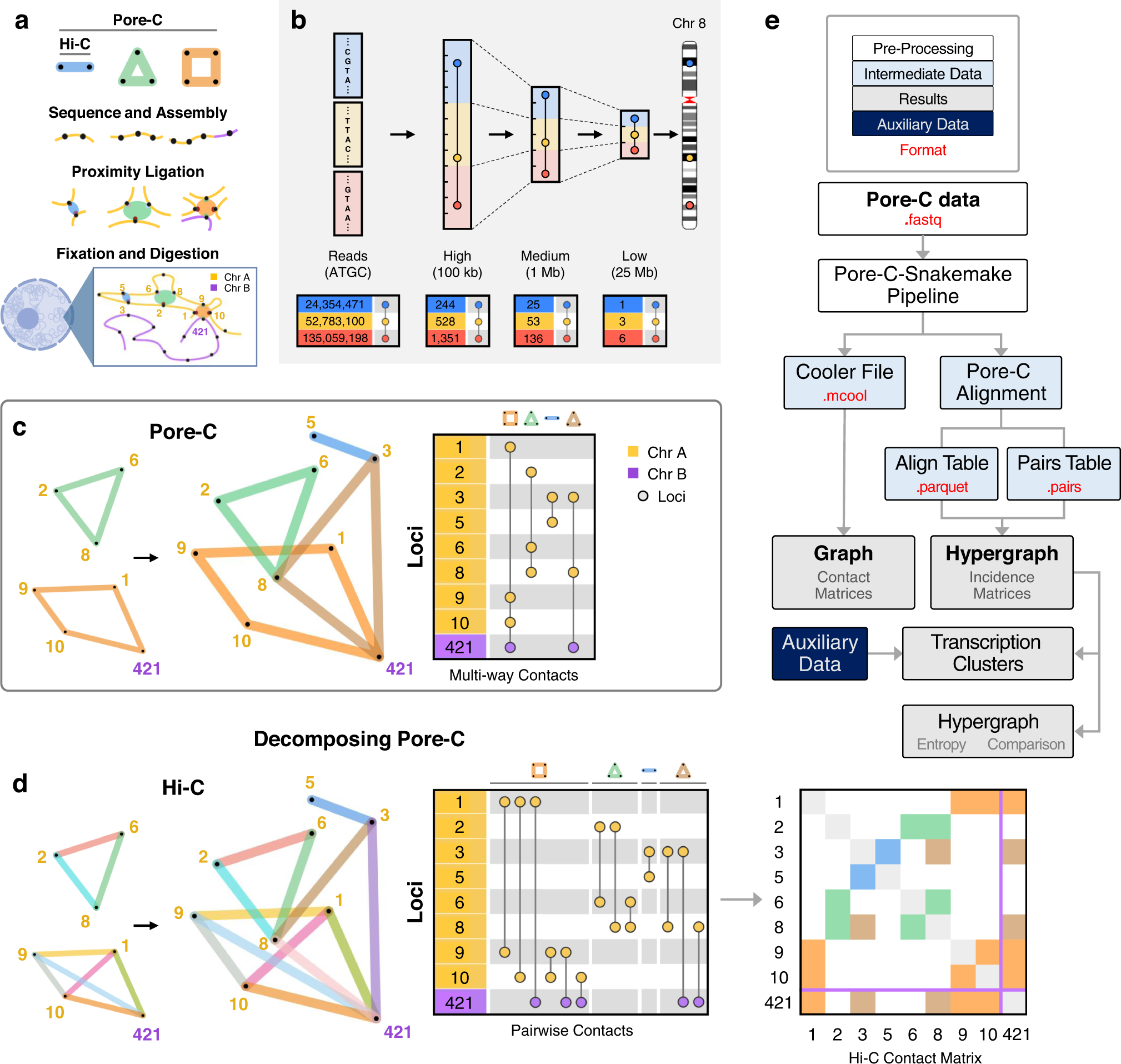
The kinetics of high-order chromatin composition is known to play a role in important biological processes such as transcription, histone modification, and methylation of DNA. Considerably, several techniques have surfaced to map these chromatin conformations. One was Hi-C, the first high throughput sequencing technique put out by Liberman- Aiden et al. in the year 2009. Researchers at the University of Michigan, to address the gaps of classic Hi-C integrated Oxford Nanotech ‘pore C’, an extension of Hi-C. Pore C is designed to analyze genomic architecture by utilizing 3C’s (Chromatin Conformation Capture) and long nanopore long sequence reads, which were earlier limited by classic Hi-C. Implementing the incidence matrix, publicly available pore C database from neonatal and adult fibroblast biopsies for B-lymphocytes was analyzed to predict novel multi-dimensional contact among distinct genome loci.
The methodology utilized a hypergraph framework, mathematically, which denotes nodes as genome loci and hyperedges as multi-way contacts. The interactions represent hypergraph entropy for different cell types. Similarly, pore C was integrated with various biological modalities to applicable interactions and termed them as transcription clusters. These clusters are believed to unfold gene identity, cell functioning, and a system of networks.
As a result, a Pore C experimental protocol using human dermal fibroblasts and neonatal fibroblasts from donors and workflow was set up to capture pairwise contact and multi-way contacts. The protocol included cross-linking, restriction digestion, and ligation of adjacent ends followed by sequencing, which allows long reads to identify and classify multi-way contacts in the genome. These multi-way contacts were further decomposed into pairwise contacts, developing hypergraphs at higher resolutions (100kb, 1Mb, 25 Mb). Within the region of chromosome 22, pairwise contacts were computed, which helped in the identification of TADs (Topologically associated Domains) using spectral identification.
Following the identification of TADs, chromosomes were constructed as hypergraphs at multiple orders, such as 2-way, 3-way, 4- way, etc., to pattern intra and inter-chromosomal contacts. Summarized multi-way contact motifs are believed to offer insights into the chromosomal architecture and their association with the regulation of transcription.
As supported by FISH (fluorescence in situ hybridization), colocalization of multiple loci can help increase the efficiency of transcription factors due to increased coordination. Since Pore C data is well suited to identify potential transcription clusters across long reads, using ATAC-seq data and selective locus binding to RNA Pol II binding based on next-generation sequencing CHIP- seq data. With the provided criteria, 12,364 neonatal fibroblasts, 16,080 adult fibroblasts, and 16,527 B-lymphocyte potential transcription clusters were identified. Data-driven identification of these transcription factors involved at least one expressed gene (94.2 % in neonatal fibroblasts, 95.0 % in adult fibroblasts, and 90.5% in B lymphocytes) with at least two expressed genes (69.6% in neonatal fibroblasts, 71.9% in adult fibroblasts, and 58.7 % in B lymphocytes).
Image Source: https://doi.org/10.1038/s41467-022-32980-z
As a result, researchers identified the colocalization of expressed genes within these transcriptional clusters. Upon identification, over half of these clusters contained multiple expressed genes that had common transcription factors (61.9% in NF, 65.2% in AF). More than half of these transcription regulators were master regulators (55.9 % in NF, 63.4 % in AF). While around 50% of clusters in B lymphocytes had multiple expressed genes with common transcription factors and a predominant percentage of 46.8% being master regulators.
To understand the chromatin architecture and transcription regulation based on these transcription clusters and master regulators, these clusters were aligned, and CCCTC- binding factor (CTCF) for binding using CHIP-seq data was evaluated. Comparing the two random multi-way contact data and identified transcription clusters, CTCF binding was higher in identified clusters. Identifying RAD21 and SMC3 cohesion subunit colocalization also preferred transcription clusters.
The data, hence, suggested transcription clusters are important sites of transcription and regulation, supporting the model in which CTCF and cohesion subunits dynamically mediated multi-way interaction.
To govern cell-type specific chromosome network architecture, these transcription factors were ranked based on the expressed frequency of binding sites. 39 % of sites were shared across the three cell types, compared to 72 % among neonatal and adult fibroblasts. Due to the overlapping of transcription binding sites across the cell types, the research team hypothesized transcription loci as a subset of these clusters, defining them as self-sustaining transcription clusters. Stratifying these clusters with stronger and weaker coupling based on transcription factor as gene analog or not. Hence, these self-sustaining transcription clusters with strong coupling act as ‘core clusters,’ serving as transcriptional signatures for each cell type.
Final Thoughts
The study extends the application of hypergraphs with mathematically simple and computationally relevant data to map and analyze genome organizations. The research team has provided biologically relevant multi-way contacts by studying transcription factors, transcription clusters, and cell type-specific chromatin architecture across cell types. The future directions supplemented by the author to explore this phenomenon systematically include time series interactions, cellular and differentiation reprogramming, and single-cell observations.
Article Source: Paper Reference | Code Availability: Pore-C_Hypergraphs | Pore-C
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Mahi Sharma is a consulting Content Writing Intern at the Centre of Bioinformatics Research and Technology (CBIRT). She is a postgraduate with a Master's in Immunology degree from Amity University, Noida. She has interned at CSIR-Institute of Microbial Technology, working on human and environmental microbiomes. She actively promotes research on microcosmos and science communication through her personal blog.





{kind=link}