
A team of computer scientists from the City University of New York has created a novel framework named the context-aware deconfounding autoencoder, which is capable of predicting the drug response elicited in a patient from a neural network model trained using cell-line data, thereby suggesting the model’s potential application in personalized medicine.
Omics profiling has shown potential utility in precision medicine. It can aid one in detecting gene or protein expression changes over time and with varying drug dosage levels. Contrasting with genomic studies, transcriptomic analyses focus on gene expression levels. Transcriptomics studies the complete mRNA transcript set present in a given cell and the quantity of each transcript. It is a powerful technique to characterize a cell’s activity and develop machine-learning models to find compounds for unique/personalized phenotypes.
During the early stages of drug discovery, cell lines and other in vitro models are extensively screened for effective drug candidates. When translated to a clinical setting, the candidate drug shows poor efficacy in patients. Therefore, to address this gap, a computational model that employs data from a panel of drug responses elicited in a patient against a particular drug candidate could prove helpful.
But, the out-of-distribution (OOD) problem poses a challenge in predicting patient-specific drug responses using machine learning. With the assumption that the data distributed on the training data and the unknown testing data are the same. The patient sample already has a limited amount of information using a machine learning model can also deteriorate its overall performance.
Another question to be addressed is how to remove biases, disentangle, extract and align common drug response biomarkers from a set of expressed genes, only using in vitro cell line models to accurately predict the best possible drug candidates with significant efficacy that has never been tested in patients yet.
Thus, to address these questions, the authors proposed a context-aware deconfounding autoencoder (CODE-AE). CODE-AE is a self-supervised training model screened for 59 drugs in 9,808 cancer patients to discover novel anti-cancer therapeutics and disease-specific biomarkers.
Findings
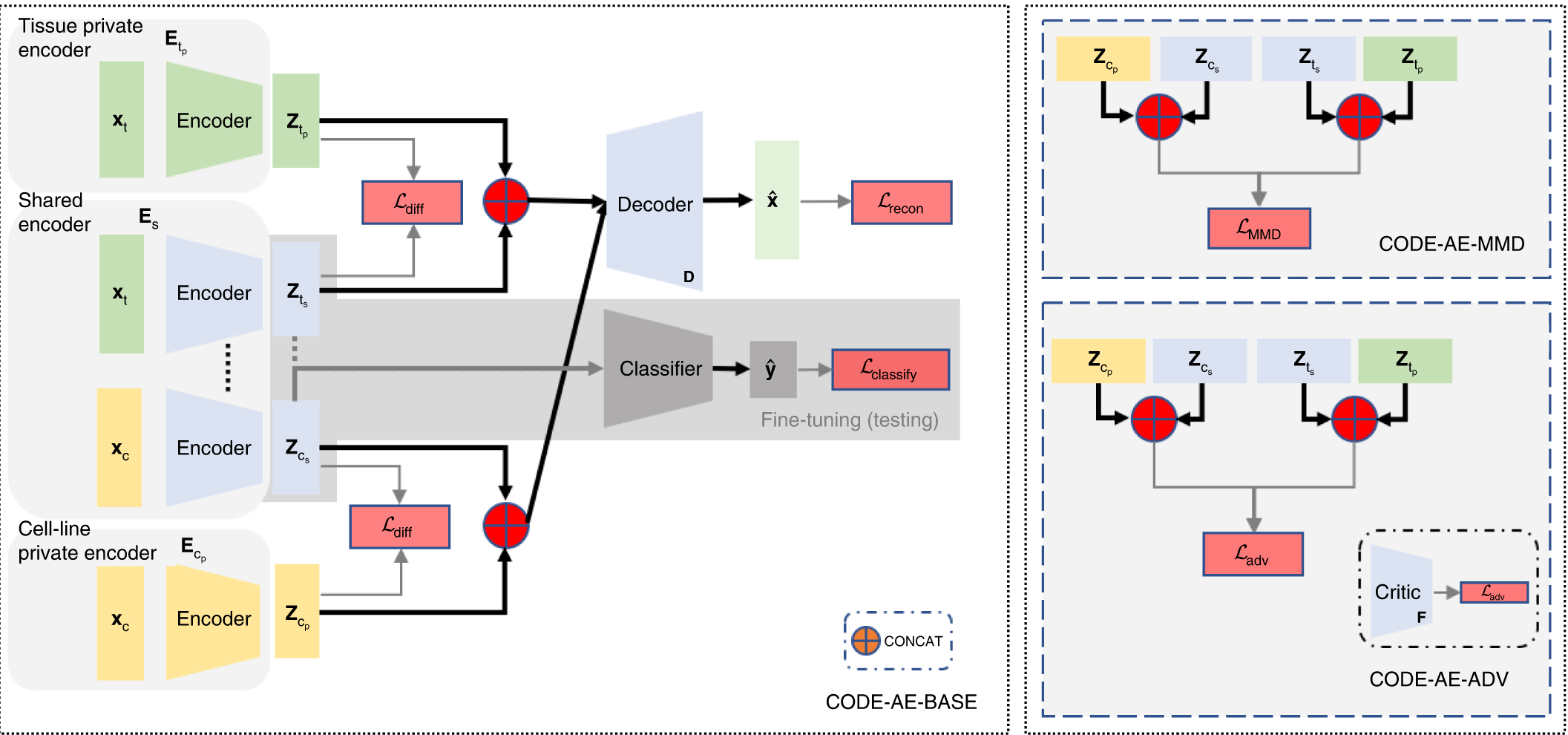
The CODE-AE architecture follows a pre-training fine-tuning approach. During the pre-training, CODE-AE collects unlabelled data from the source (cell lines) and the target (patient sample), thereby training the autoencoder.
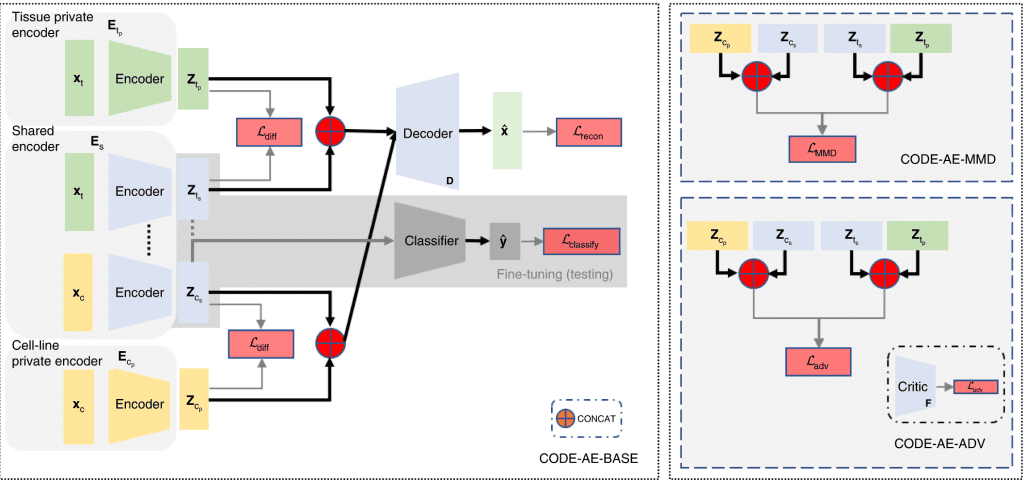
Image Source – https://doi.org/10.1038/s42256-022-00541-0
CODE-AE possesses two novel features,
1) It learns both shared and unique signals to both cell lines and the patients, with an aim to disentangle the common biological signals.
2) CODE-AE standardizes the embeddings of cell lines and patients to make their distributions similar. Doing this can transfer the knowledge learned from the cell-line model to patients.
Next, to address the OOD problem, CODE-AE-ADV ( a regularization method that minimizes adversarial loss) was shown to effectively align cell line samples obtained from the Cancer Cell Line Encyclopedia (CCLE) with the patient dataset obtained from The Cancer Genome Atlas (TCGA). Although any autoencoder could do this separation, CODE-AE-ADV was proven more effective in transferring gene expression profiles, thereby addressing the OOD.
To determine the robustness of CODE-AE-ADV, the autoencoder was trained using the CCLE dataset to screen the efficacy of 59 drugs against 9,808 cancer patients. The cellular viability observed in various cell lines in response to the given drug is treated as the variable.
Nine thousand eight hundred eight patient data were categorized into 45 clusters with a drug response score calculated by averaging the individual patient response score. The model determined the respective z-scores for the particular drugs’ efficacy in a patient.
Patients with lung cancer, head and neck cancer, cervical and endocervical cancer, urothelial bladder carcinoma, and oesophageal carcinoma were clustered together and were searched for effective drugs. The CODE-AE-ADV showed gefitinib, 5-aminoimidazole-4-carboxamide ribonucleotide, and gemcitabine as the top 3 hits for the most responsive drugs. With gefitinib being implicated as a treatment option for head and neck cancer, undergoing clinical trials for cervical cancer, urothelial bladder carcinoma, and oesophageal carcinoma go on to show the auto encoders’ clinical relevancy.
Concluding Remarks
The pre-trained autoencoder CODE-AE-ADV, developed by He et al., successfully predicts an individual patient’s drug response by utilizing cell line data. CODE-AE can be further extended to multi-omics data instead of only transcriptomic data to determine drug efficacy. The computational framework can be improved by incorporating biological pathway information and gene ontology data. Although this study focused on the accuracy of CODE-AE in precision oncology, the model can very well be used to train other disease models in the future.
Article Source: Reference Paper | Code Availability: Zonodo | CodeOcean
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Shwetha is a consulting scientific content writing intern at CBIRT. She has completed her Master’s in biotechnology at the Indian Institute of Technology, Hyderabad, with nearly two years of research experience in cellular biology and cell signaling. She is passionate about science communication, cancer biology, and everything that strikes her curiosity!





{kind=link}