
Scientists from the Duke University with collaborators at the University of Connecticut are aiming to create novel enzyme inhibitors to combat Methicillin-resistant Staphylococcus aureus (MRSA). The researchers revealed how a single small mutation affects therapeutic efficacy in a study published in PLOS Computational Biology.
Antimicrobial resistance is an important public health issue worldwide. New resistance mechanisms continue to evolve, posing a threat to the efficacy of already available medications. In several situations, antifolate resistance has arisen, including resistance to antifolate trimethoprim (TMP), the sole FDA-approved DHFR inhibitor for antibiotic usage. As a result, second-generation antifolates must be created to combat resistance. This requirement drives the research, which aims to create and utilize fast and accurate computational approaches for predicting and understanding resistance processes. MRSA is a bacterial infection that has developed resistance to most antibiotics used to treat common staph infections.
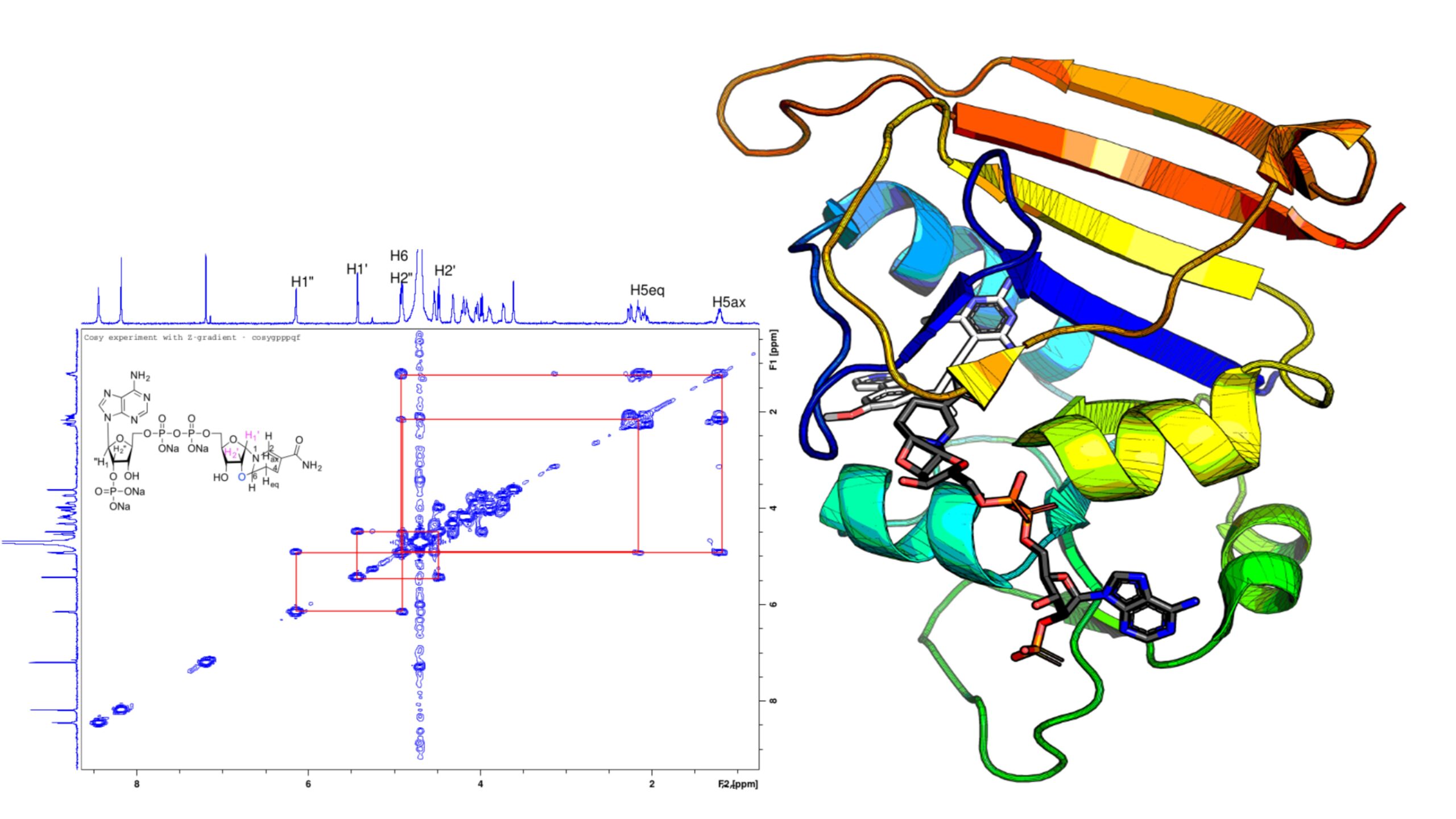
The study led by Bruce Donald, Distinguished Professor of Computer Science at Duke University, looked at dihydrofolate reductase (DHFR), an enzyme targeted by antibiotics to combat MRSA. Inhibitors of DHFR act like locks and keys, binding to MRSA enzymes that have a unique three-dimensional structure that only permits molecules that fit perfectly to adhere to them. Antifolates can be created as selective and safe antibacterial drugs due to structural and sequence differences between human and prokaryotic DHFR.
A bacterial enzyme’s structure can be altered by a mutation, rendering medicines ineffective. A well-known resistance mutation is the F98Y mutation. A little alteration in the DHFR enzyme’s 98th amino acid converts phenylalanine to tyrosine. “Those two amino acids are structurally similar,” said Graham Holt, a Donald lab graduate student, “but the mutation has a huge effect on the efficacy of the inhibitors.” It essentially alters the lock.
Pablo Gainza, Ph.D., a former graduate student in the Donald lab, believed that using OSPREY, a set of programs for computational structure-based protein design developed in the Donald lab, he should be able to predict this mutation. However, he was unable to do so. He went back to investigate the beginning structure after knocking down hypothesis after hypothesis to figure out why he couldn’t foresee this mutation.
“We looked at the electron density data from the crystallographer and found something strange,” says Donald.
Crystallographers utilized a computer software that, unknown to them, inverted the chirality, or produced a mirror image, of the NADPH cofactor to gain a better match while trying to figure out the structure of the F98Y mutant. They determined that the “flipped” chemical species they uncovered via their investigation existed under laboratory circumstances and is likely to exist in vivo.
“Using OSPREY, we discovered this flipped chirality,” said Donald, “which we believe happened because of the F98Y mutation.” Just as in 2-factor authentication, the single enzyme mutation and the flipped cofactor appear to operate together to get around the inhibitor.
The fundamental basis for resistance is altered by this “chiral evasion.” But now Donald and his colleagues know not just how a single little mutation altered the lock, but also the structure needed to create a better key — a better pharmacological inhibitor.
“This is the first example of an enzyme that exploits the chirality of its cofactor in order to evade its inhibitors,” said Holt. “Now that we see this happening, that will help inform computational strategies to develop better inhibitors.”
It was demonstrated in the Donald lab that when flipped chirality is taken into consideration, OSPREY’s predictions closely match experimental inhibitor potency measurements. The collaborators at the University of Connecticut performed biochemical studies to test the notion and give structural support.
“This is only the beginning of the story,” Donald said. “Our discovery of chiral evasion should lead to more resilient inhibitors: better drug designs.” Most drug development is currently reactive, waiting for resistance to emerge, which it always does. “We hope to make drug design proactive, by using our algorithms to anticipate resistance,” Donald said.
Story Source: Wang S, Reeve SM, Holt GT, Ojewole AA, Frenkel MS, Gainza P, et al. (2022) Chiral evasion and stereospecific antifolate resistance in Staphylococcus aureus. PLoS Comput Biol 18(2): e1009855. https://doi.org/10.1371/journal.pcbi.1009855
Background Image Source: https://genome.duke.edu/news/wed-02162022-0809/looking-mirror
Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.





.){kind=link}