
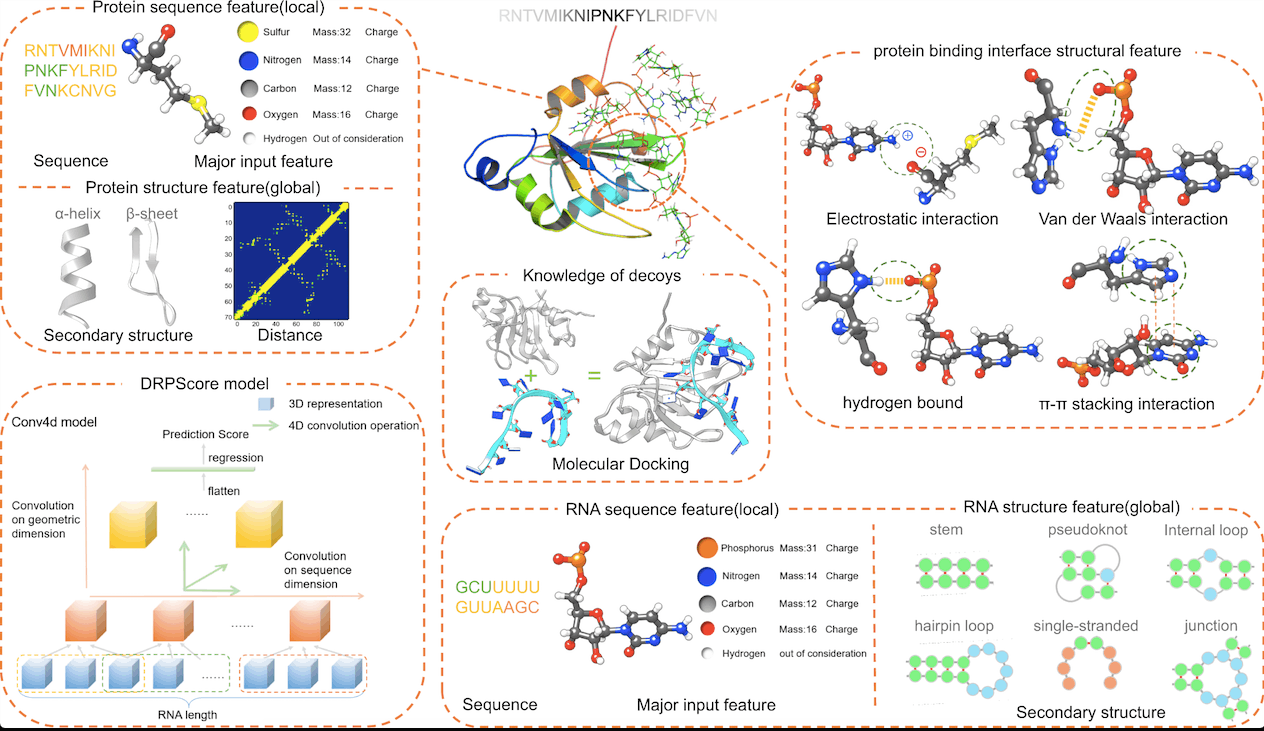
Scientists have developed a new deep-learning-based approach called DRPScore for identifying native-like RNA-protein complexes essential for understanding cellular processes. DRPScore outperforms existing methods in identifying native-like structures with high accuracy, even in challenging cases involving significant conformational change. The method was tested on sets of RNA-protein complexes with varying degrees of conformational change induced by binding, from fully rigid to fully flexible docking.
Limitations of previous approaches and DRPScore
RNA-protein complexes have vital roles in a variety of biological processes and diseases. Unfortunately, because of technological constraints, experimental identification of their three-dimensional structures is difficult. To anticipate and assess RNA-protein complex architectures, computational approaches, including propensity-based and atomic-level statistical scoring functions, have been developed. Propensity-based scoring functions statistically evaluate the interface propensity of paired nucleotide residues and derive a possible formula from the inverse Boltzmann formula.
DARS-RNP is a coarse-grained propensity-based scoring system that employs a simplified representation of protein and RNA to generate the scoring function using four terms: steric clash penalty and distance, angle, and site dependencies. Yet, it is still difficult to accurately predict the architectures of RNA-protein complexes because of the limited scoring functions available.
To help with this understanding, computational approaches for predicting the structure and binding affinity of RNA-protein complexes have been devised. Consideration of structural flexibility and conformational changes upon binding is one of the primary problems in this field.
In their paper, Xiao et al. suggested a unique scoring function, 3dRPCScore, based on statistical potential energy. This method proved superior to propensity-based scoring systems, which cannot account for conformational changes.
Atomic-level statistical scoring functions, which employ distance-dependent interaction potentials that follow the Boltzmann distribution, were also used. Unfortunately, all these approaches struggle with unbound docking.
Xiao et al. presented DRPScore, a deep-learning-based RNA-protein complex scoring function that takes structural flexibility into account. The rigorous evaluation of DRPScore on RNA-protein testing sets revealed significant improvements over the previous approaches.
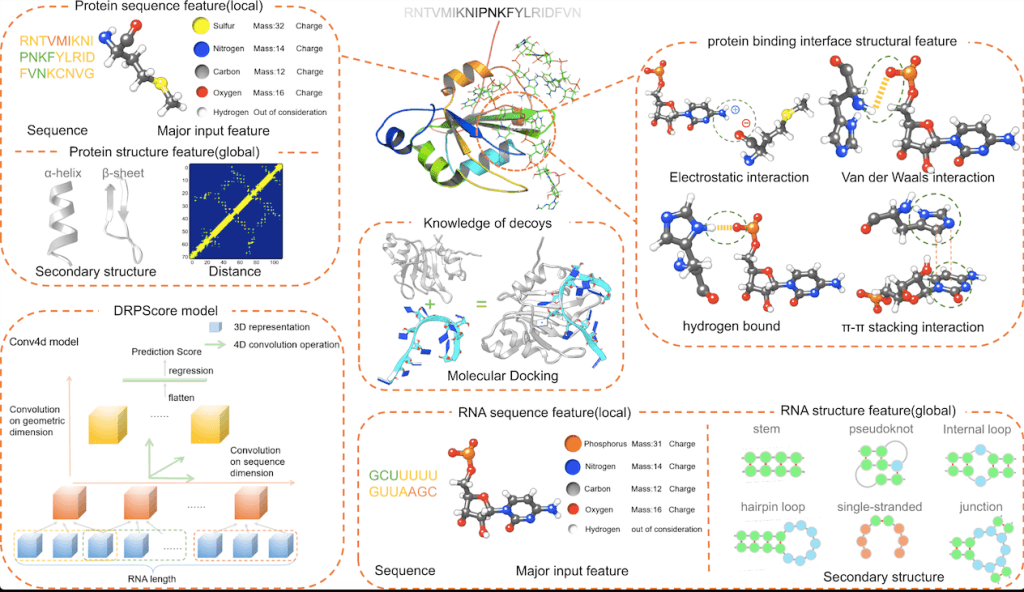
Image Source: https://doi.org/10.1038/s41467-023-36720-9
Bound RNA-protein testing sets
Four different computational methods, DRPScore, ITScore-PR, DARS-RNP, and 3dRPC, were evaluated for predicting the structures of bound RNA-protein complexes. The evaluation was based on three factors: the success rate of predictions, ranking, and Irmsd (a type of root mean square deviation).
In terms of prediction success rate, DRPScore defeats the other three approaches, with an average success rate of 80.56% for the top 5 predictions and 91.67% for the top 20 predictions. This indicates that DRPScore can precisely identify the native-like bound-bound RNA-protein structures. Overall prediction ranges, DRPScore consistently performs better than the other three approaches.
Unbound RNA-protein testing sets
The researchers used either homologous modeling or other unbound RNA-protein complexes to obtain at least one partner of each unbound RNA-protein complex. They compared the performance of DRPScore with three other well-known scoring functions, namely ITScore-PR, DARS-RNP, and 3dRPC. The evaluation was based on the top 5 and top 20 predictions, and the success rate was measured as the fraction of forecasts with RMSDs less than 8. The predictions were categorized into four groups based on RMSD ranking.
The results showed that DRPScore consistently outperformed the other three scoring methods in predicting both bound and unbound RNA-protein complexes. DRPScore had a higher proportion of accurate predictions and a lower percentage of predictions with high RMSD. Additionally, DRPScore was more precise in recognizing RNA-protein interface features when there were a limited number of interface contacts.
Local and global property analysis of interface interactions
DRPScore can infer interface interactions, including the detection of hydrogen bonds and secondary structure interactions, by evaluating both local and global properties such as α-helix and β-sheet for proteins and hairpin loop, internal loop, bulging loop, junction, and pseudoknot for RNA. DRPScore found a greater proportion of hydrogen bonds and loop-helix secondary structure interactions at the RNA-protein interface than previous statistical potential functions-based approaches, resulting in a more precise evaluation of RNA-protein complexes.
Comparison to traditional deep-learning methods
Intra- and inter-nucleotide/residue interactions are captured by DRPScore, which uses a 4DCNN model to extract and transfer 3D coordinates information to pictures and captures intra- and inter-nucleotide/residue interactions. Traditional 3DCNN models, on the other hand, only collect intra-nucleotide/residue information and disregard inter-nucleotide/residue interactions, resulting in reduced accuracy in recognizing native-like RNA-protein complexes. Model training for DRPScore employs a back-propagation-based mini-batch gradient descent optimization approach. DRPScore can analyze 1000 RNA-protein complex structures in around 8 minutes. Comparing DRPScore, 3DCNN, and other approaches revealed that DRPScore identifies native-like RNA-protein complexes with a greater success rate.
Conclusion
Despite the fact that their deep learning-based model, DRPScore, surpasses conventional scoring functions in RNA-protein structural assessment, the researchers highlight that there is still potential for advancement. Prior attempts in this sector particularly neglected structural flexibility in favor of rigid-body docking. In the case of completely flexible unbound-unbound docking, the interaction interface may change significantly, and scoring algorithms may not have previously learned analogous structures.
To overcome this issue, the scientists propose including molecular dynamics modeling to explicitly account for the flexibility of RNA-protein complexes. This would permit the sampling of the dynamical conformations that RNAs and proteins form when interacting, which is a key bottleneck in the field of study at present. In doing so, scientists expect to enhance the precision and robustness of their model and advance the area of RNA-protein structure assessment.
Article Source: Reference Paper
Learn More:




Sejal is a consulting scientific writing intern at CBIRT. She is an undergraduate student of the Department of Biotechnology at the Indian Institute of Technology, Kharagpur. She is an avid reader, and her logical and analytical skills are an asset to any research organization.





{kind=link}