
Researchers at the Broad Institute of MIT and Harvard have developed Genebass, a free online browser that will assist other researchers and clinicians studying specific genes in linking rare genetic variants with disease phenotypes. Genebass represents a genetic analysis of nearly 400,000 people from the UK Biobank and may assist researchers in identifying potential novel therapeutic targets.
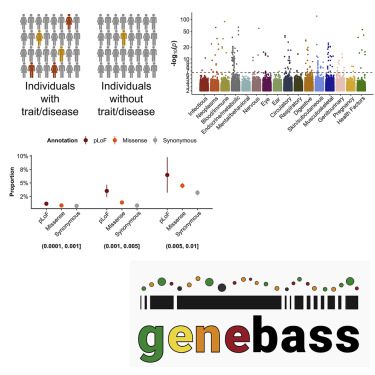
Human diseases and traits have been associated with thousands of common genetic variants found in genome-wide association studies, but rare variations in human diseases have not been studied at scale. A streamlined method of evaluating the impact of rare coding variations across a wide range of phenotypes is possible with exome sequencing of population biobanks. An association analysis was performed using exome-sequence data from 394,841 individuals in the UK Biobank on 4,529 phenotypes. Genetic associations are closely correlated with frequency and deleteriousness, as well as natural selection metrics. The dataset is released as a public resource alongside the Genebass browser for exploring rare-variant association results rapidly and highlights biological findings revealed by these data.
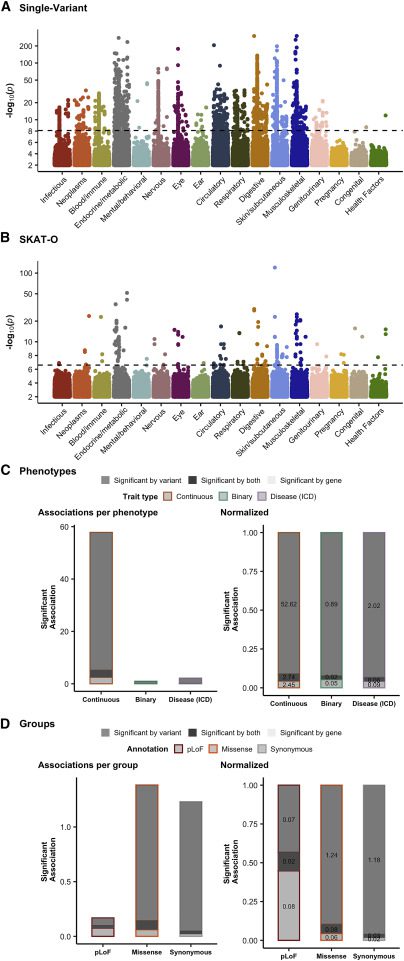
Image Source: https://doi.org/10.1016/j.xgen.2022.100168
After the development of the gene model and mapping of the human genome, coding variation has been the most readily interpretable class of genomic variation. The actionable variant list from the American College of Medical Genetics, which maps and interprets genetic variants of immediate clinical significance, has been made possible by this technology. As a result of exome sequencing, specific causal variants have recently been discovered for hundreds of rare diseases, especially for de novo dominant variants causing severe disease. It is becoming increasingly possible to identify associations between rare variants and phenotypes (both complex traits and diseases) as sample sizes grow for exome sequencing datasets.
It is possible to attain direct insight into potential therapeutic avenues for complex diseases by identifying causal genetic factors. Increasing low-density lipoprotein levels (LDL), for instance, has been demonstrated to increase cardiovascular disease risk due to variants in PCSK9. A therapeutic approach to inhibit PCSK9 has been brought to market less than 15 years after loss-of-function variants were found to prevent cardiovascular disease. Using deeply phenotyped biobanks, it is possible to simultaneously analyze multiple diseases and traits within a single cohort, enabling the identification of rare variants of ANGPTL7 that protect against glaucoma, for example. This approach facilitates the discovery of new disease genes with therapeutic potential.
A genome-wide association study (GWAS) has been extensively conducted on the UK Biobank, which holds standardized and detailed phenotypic data on about 500,000 participants. In collaboration with biopharma companies, the UKB Exome Sequencing Consortium generated exome sequences for this cohort.
Exome sequence data has been used in recent studies to examine various aspects of rare-variant associations. There have been new hits for a variety of traits in cross-phenotype analyses, as well as new biochemical signals for type 2 diabetes and cardiometabolic traits. The goal of this study was to perform a systematic, large-scale analysis of associations between rare variants and phenotypes. The results are released in a results browser, along with a discussion of the role of natural selection and allele frequency in the analysis.
A public-facing Genebass browser (https://genebass.org) and bulk data downloads are available to the public for 4,529 phenotypes for rare-variant association analysis. In this article, SCRIB’s association with a brain imaging trait as well as its correlation with natural selection, allele frequency, and genetic discovery is discovered. To determine the extent and role of pleiotropy among rare variants, as well as their contribution to the heritability of common diseases, future work will be needed.
Research limitations:
The analysis that was conducted had a number of limitations. However, even on a scale of 400,000 individuals, it was found very small associations for many diseases, even with extensive QC to improve reliability. Due to this, we urge caution when interpreting association results, particularly when the data counts are small (asymptotic properties) for the rarest binary traits and ultra-rare genetic variants.
Increased numbers of cases are important for analyzing rare coding variations across genes in the presence of the rarest outcomes. Firth regression may also be effective for such traits as an alternative to other statistical methods. Despite this, mixed-model tests for rare binary traits seem to have poor asymptotic properties since lambda GC for these tests drops precipitously as the number of binary traits increases. In addition, genes marked by phenotypic associations are clearly enriched for natural selection at the gene level, such as LoF-intolerant genes.
Furthermore, these association analyses were restricted to individuals who have European descent, the largest group in the dataset. The power and resolution of genetic discovery are enhanced as additional ancestries are added to these analyses. As the UK Biobank has a very limited sample size for non-European individuals, most binary traits, including most disease outcomes, would be underpowered. For more insight into the contribution of rare variants to common disease etiology, large biobanks with diverse participants are needed to overcome these limitations.
Article Sources: Research Paper | Reference Article
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Srishti Sharma is a consulting Scientific Content Writing Intern at CBIRT. She's currently pursuing M. Tech in Biotechnology from Jaypee Institute of Information Technology. Aspiring researcher, passionate and curious about exploring new scientific methods and scientific writing.











{kind=link}