
Witnessing the most wrecking pandemic in a decade, COVID-19 visibly tested the potential of healthcare advancements. A collaborative team of researchers addressed earlier loopholes in research by systematically identifying protein interaction contacts between humans and the sars-cov-2 proteome. The team successfully evaluated the ‘contactome theory’, associating the host genome with morbidity and the COVID-19 community network.
Introduction
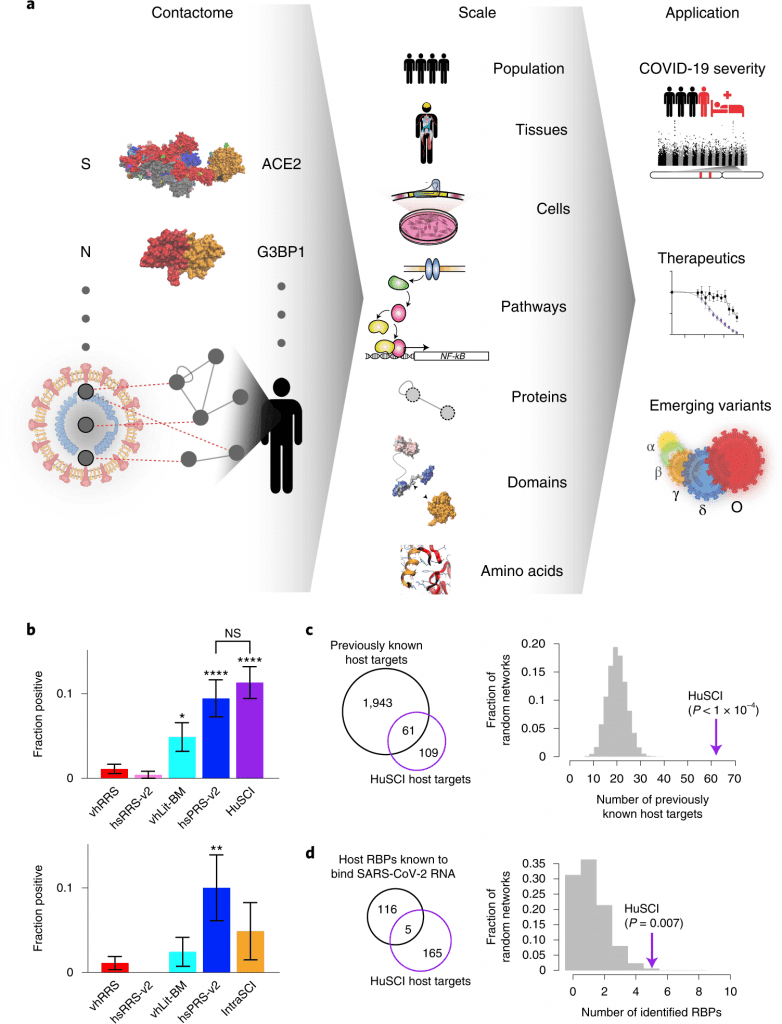
Image Source: https://doi.org/10.1038/s41587-022-01475-z
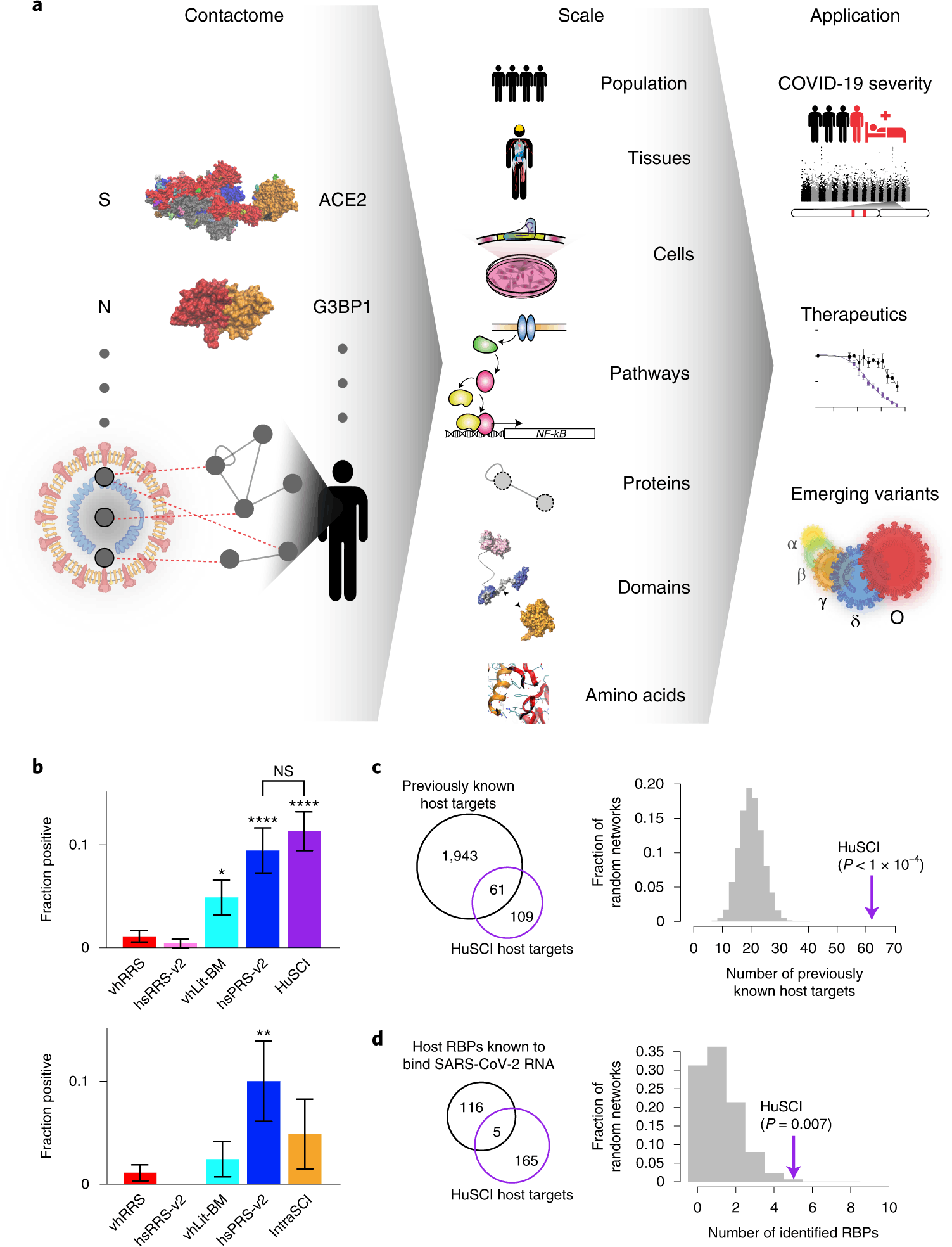
The COVID-19 pandemic reasoned the perspective of healthcare and individual health of individuals. The possibility of restoring the new normal was among the highlights of the research, it largely helped to bridge comorbidities and reasons for them. While the preliminary research and vaccine focussed on the ACE-2 receptor and spike proteins of the sars-cov-2 virus, the other proteome factors of both the host and virus were largely ignored. To address this gap, a collaborative effort of researchers from the University of Toronto, Harvard Medical School, Aix-Marseille Université, and Ludwig-Maximilians-Universität (LMU) by bridging infection and immune-mediated interaction. These interactions between the viral molecules and human proteome and identify the virus-host contacts, also referred to as ‘contactome,’ were also identified. The team successfully mapped the genetic architecture of the disease and potential therapeutic targets.
The methodology used a multi-assay and screening to generate a high-quality virus-host contactome map. In order to increase the sensitivity, the framework used a yeast two-hybrid tool (Y2H) and used two complementary versions of the assay. The first version was a plate-based version using ‘balt’ and ‘prey’ fusion proteins with N-terminal and GAL1-HIS3-based growth selection (Y2H-HIS3), and the second version included a new system based on barcode fusion genetics (BFG)-Y2H using C-terminal prey protein expressing GFP protein (Green Fluorescent protein).
The human interactors were pairwise retested in triplicate against each variable open reading frame. The yielding 118 interactions involved a total of 14 viral and 92 human proteins. These Y2H-HIS3-based human sars-cov-2 interactome datasets were referred to as HuSCI-HIS3. Upon mapping the sars-cov-2 host contactome, a total of seven interactions were identified in both HuSCI-GFP and HuSCI-HIS3. The overlapping is consistent throughout the complementary nature of the assays and pipeline.
The secondary direction was to determine the complementarity of contactome and co-complex datasets by investigating host and viral proteins of covid-19 causing sars-cov-2 using affinity purification followed by mass spectrometry to identify complex associations and biotin identifications (BioID) to determine proteome architecture. In order to identify the co-complex associations, contacts were computationally modeled. In both BioID and co-complex studies for the dataset, 20 out of 204 Hu-SCI interacting pairs were identified, and approximately 34% were associated with both host and viral contactome.
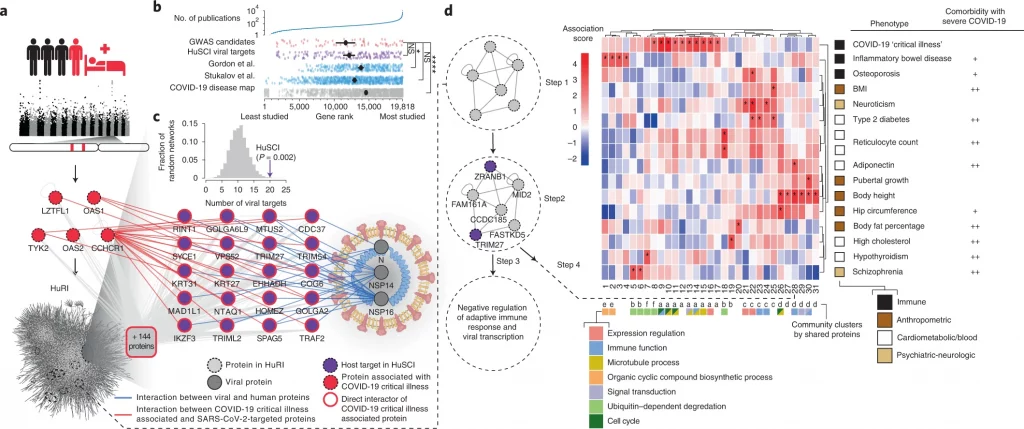
Image Source: https://doi.org/10.1038/s41587-022-01475-z
The research team defined both ‘tissue-specific’ and ‘common’ human proteins using the Human Protein Atlas (HPA). Both spectrometry and proteome identification results were biased towards the common host proteins and also exhibited protein expression in the diverse tissues, which essentially showed infection of sars-cov-2 due to their ability to have specific tissue disruption.
In order to identify the targeted function and affected pathways by the covid-19 causing virus. The broad function of host proteins, such as mediation of immune responses, the infectivity of viral process, ubiquitination of protein, cytoskeleton formation, and protein transport across the cell organelles using vesicle-mediated transport. Scientists focussed on identifying pathways largely associated with inflammatory responses and viral proteins in mediating infections. The key regulators from the viral proteome were identified as NSP9, NSP14, and NSP16 in mediating cytokine responses such as REL (NF-κB subunit) and IKBKG (inhibitor of NF-κB kinase regulatory subunit). These results mediated results for direct protein interaction function within the immune pathways and were of relevant inference to disease-causing sars-cov-2.
In order to identify the severity linkage of COVID-19 symptoms and outcomes. The underlying mechanism in response to human proteome was analyzed using molecular mechanisms. Previous meta-analyses in recent studies identified the linkage of genetic loci with severe COVID-19. Hence, the researchers utilized the two models to conceptualize in reference to genetic variations. The two models were:
1) Direct Model: In this model, genetic variation relates to virally targeted host proteins, and these modulate the disease outcome. Alternatively,
2) Indirect Model: In this model, genetic variation in the network neighborhood of targeted host proteins modulates the downstream effects, eventually adding to disease outcomes.
The results identified the direct model was not supported due to no targeted host protein from HuSCI being encoded from severe illness and critical condition. Whereas there was consistent with the indirect model, proving clinically relevant genetic variation acts in the local network neighborhood of viral contact proteins in the binary host network maps.
Final Thoughts
Evaluating the impact of emerging viral strains throughout the COVID-19 pandemic sustained the research to spike protein and ACE2 receptor interactions. The systematic contactome mapping of coronavirus SARS-CoV-2 alongside the human host encompassed more than 200 binary virus-host and intra protein-protein interactions providing an opportunity to connect genes and their architecture. The potential of the viral nucleocapsid proteins in the host showed the emergence and potential targets other than ACE 2 receptors, understanding severity and overcoming the effect of variants.
Article Source: Reference Paper | The protein-protein interaction (PPI) data: IMEx | Code availability: GitHub
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Mahi Sharma is a consulting Content Writing Intern at the Centre of Bioinformatics Research and Technology (CBIRT). She is a postgraduate with a Master's in Immunology degree from Amity University, Noida. She has interned at CSIR-Institute of Microbial Technology, working on human and environmental microbiomes. She actively promotes research on microcosmos and science communication through her personal blog.





{kind=link}