
Japanese scientists from the National Institute of Natural Sciences (NINS) and Osaka University have discovered novel protein folds. They created a well-defined set of rules to predict the structure of unprecedented αβ-folds and then designed those structures through de novo design. All eight predicted structures match the de novo design models. They were also able to create a knot-like αβ fold, which was previously thought to be impossible. The implications of this finding are profound, suggesting that a vast number of αβ-folds are theoretically possible but have either not emerged or have become extinct due to evolutionary bias. This research could have far-reaching consequences for understanding protein evolution and its potential applications in various fields, including biotechnology and medicine.
Exploring the Proteins “NOT” Found in Nature
From structural roles to immunological ones, proteins perform various functions vital to our body. And in the case of proteins, the three-dimensional structural diversity is strongly related to functional diversity. An important factor in determining protein structure is its fold which refers to the three-dimensional arrangement and associations between the secondary structural elements. Numerous types of folds have been discovered by solving the protein structures of many proteins found in nature.
But, over the recent years, the discovery of new types of folds has become rare. While it is logical to assume that the rare discovery of novel folds indicates that all the possible types of folds have been uncovered, some scientists believe that unique folds have either been overlooked, have not yet appeared in nature, or have existed but have since gone extinct due to natural selection.
To determine whether this hypothesis holds true or not, a team led by Nobuyasu Koga designed folds de novo that are not present in nature. Till now, exploration of fold space has been done only on naturally occurring folds, except for a novel fold, Top7. To go beyond naturally occurring folds, the researchers developed certain rules regarding how β-sheets (type of protein secondary structure) can be arranged in new and unique yet feasible ways. These rules serve as a “guide” for designing unprecedented αβ-folds.
The World of αβ-folds
More than 50% of the folds in nature belong to αβ folds. These folds are characterized by a specific arrangement of β-sheets, and most of them are crucial in the functioning of enzymes. In this study, the researchers sought to ascertain whether there exist any undiscovered αβ-folds in which the β-sheets have three to eight strands arranged in an open conformation without forming a barrel shape.
For this purpose, they focused on patterns in which β-strands follow Richardson’s rule. This rule states that the connections between parallel-aligned β-strands should have a right-handed orientation. Based on this criterion, they found a lot of potential αβ-folds, many of which do not occur naturally. But among all these folds, some were just not possible in reality, such as the one with overlapping connections between the β-strands. To address this issue, they devised a set of rules for β-sheet topology to predict the αβ-folds that are likely to be possible. The rules they came up with are listed below:-
- Connection jump-distance rule:- As per this rule, if two β-strands have a large number of β-strands between them, the arrangement is not favorable. The jump distance between parallel β-strands should be less than four, and that for antiparallel β-strands should be less than two. The only exception is Greek key topology (an antiparallel motif), where two intervening β-strands are allowed.
- Connection overlap rule:- An overlap between the connections of two β-strand motifs is not favorable. S-type β-sheet topologies (connections on the same side) are less preferred than D-type ones (connections on different sides).
- Connection ending rule:- If the second strands of two parallel β-strand motifs are adjacent and aligned in a parallel manner, S-type topologies are favored over D-type ones.
Fascinating Findings from De Novo Designed αβ-folds
Based on the aforementioned rules, the researchers grouped all open β-sheet topologies with three to eight strands into two groups, frustration-free (the ones that don’t violate the rules) and frustrated (the ones violating the rules). Many of the observed αβ-folds belonged to the frustration-free group, and the ones not commonly observed were a part of the frustrated group. Also, the frustration-free folds belonged to more homologous groups (evolutionarily independent groups).
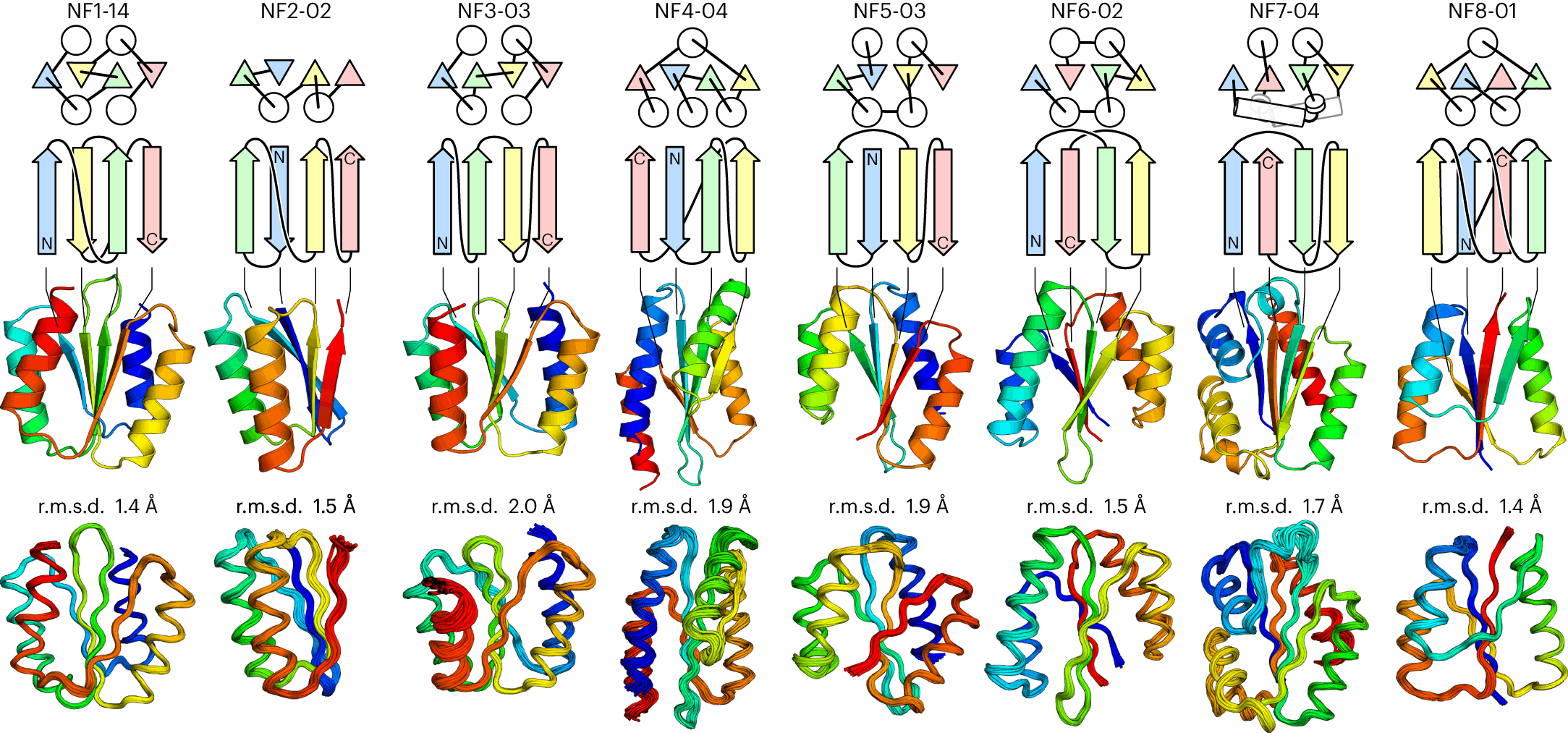
In the case of four stranded αβ-folds, from a total of 96 patterns, 43 were frustration-free. Out of those, 35 were found in nature. The remaining eight were either unobserved or rarely observed in nature, which indicates that they may not be robustly foldable. Surprisingly, one of the eight frustration-free structures forms a knot that was earlier considered impossible to exist in nature. Still, the researchers selected all eight structures for de novo design.
From Concept to Reality: Designing Novel αβ-folds
The researchers then designed the eight frustration-free structures with de novo techniques and named them NF1 to NF8. They first developed backbone blueprints for each of them, specifying secondary structure lengths and loop torsion patterns to favor the target fold. In some cases, additional α-helices were added to enhance the hydrophobic cores of the proteins.
Then, a backbone structure was created for each blueprint by multiple structures generated through simulations. They analyzed existing databases to determine whether similar structures existed and found that similar structures did exist but only for NF2 and NF4. Next, a design algorithm was implemented to add side chains (specific amino acids) to the backbone. Designs with low energy, tight packing of the protein core, and high compatibility between the local sequence and structure were selected.
Finally, designs exhibiting favorable energy profiles were experimentally characterized. For characterization, synthetic genes for all the predicted αβ-folds were obtained and used to produce the proteins using Escherichia coli bacteria. The proteins were purified and subjected to tests such as CD (Circular Dichroism) spectroscopy, size exclusion chromatography, and NMR spectroscopy.
56 proteins out of the 60 designs were successfully produced and were soluble. They exhibited characteristics of αβ-folds in their CD spectra, had well-dispersed NMR peaks, and showed high thermal stability. However, the thermal denaturation curves of some designs (N3 to N6) were not cooperative, which may be why they have not been found in nature.
The researchers also expanded their evaluation to αβ-folds with five to eight β-strands. They found that, as the number of β-strands increases, the number of possible frustration-free structures that are not yet observed also increases exponentially. They identified 12,348 frustration-free structures with five to eight strands that are not observed in nature. This is much larger than the number of αβ-folds (400) that have actually been observed in nature.
Conclusion
The observations obtained from the study indicate that the devised rules were successful in the accurate prediction of novel folding patterns. This means that over 10,000 new types of protein folds are possible but have not been found in nature yet. Additionally, researchers implemented a deep learning method called AlphaFold2 for predicting structures based on amino acid sequences. Even though it proved to be a challenging task, the fact that novel protein folds are feasible opens up exciting new possibilities for the artificial synthesis of proteins with desired functions and their applications in various fields of science and technology.
Article Source: Reference Paper
Learn More:




Neegar is a consulting scientific content writing intern at CBIRT. She's a final-year student pursuing a B.Tech in Biotechnology at Odisha University of Technology and Research. Neegar's enthusiasm is sparked by the dynamic and interdisciplinary aspects of bioinformatics. She possesses a remarkable ability to elucidate intricate concepts using accessible language. Consequently, she aspires to amalgamate her proficiency in bioinformatics with her passion for writing, aiming to convey pioneering breakthroughs and innovations in the field of bioinformatics in a comprehensible manner to a wide audience.





{kind=link}