
Scientists from Shaanxi Normal University, China, developed a novel Drug-Drug-Interactions (DDI) prediction tool that implements a dual graph neural network architecture to incorporate chemical substructure information as well as substructure interaction information of drugs. This never-before-implemented approach has the potential to identify synergistic drug combinations for COVID-19.
Why do we need to predict DDI?
Drug-Drug Interactions (DDIs), the interactions between drugs at the molecular scale, are crucial in the field of drug delivery. This is because interactions between multiple administered drugs can lead to adverse effects in patients. DDIs are also known to alter the metabolism of each drug involved, leading to reduced efficacy or toxicity. On the other hand, several synergistic drug combinations are known to increase drug efficacy and reduce toxicity. This imposes the necessity of developing advanced methods for DDI predictions.
Determining molecular representations from molecular structures is crucial in designing drug discovery methods. SMILES (simplified molecular input line entry specification) is a linear molecular representation. The representations of molecular topologies are governed by chemical rules. However, Graph Neural Networks (GNN) using graphical representations of molecules based on their geometry have surpassed the linear molecular representation approach. Drug-drug interaction (DDI) prediction is an application of molecular representation.
According to the similarity hypothesis for drugs, if drugs A and B interact to produce a certain kind of biological effect, then drugs similar to drug A would behave likewise with drug B and vice versa. The similarity between drugs is typically determined based on similar chemical structures, targets, pathways, and individual side effects. Several methods exist for predicting DDI based on the similarity hypothesis. However, these methods do not consider the individual molecular structure and interactions between chemical substructures of the concerned drug molecules.
Machine learning aided DDI: previous methods pave the way
With the rapid onset of machine learning-based tools in drug discovery, DDI prediction has also witnessed the development of a plethora of deep learning-aided methods. The DDIMDL method is based on a multimodal deep learning framework incorporating diverse drug features predicting DDIs. DPDDI used a deep feedforward neural network fed with latent drug representations learned using a deep graph auto-encoder.
The method DANN-DDI integrates several drug features effectively using a deep attention neural network framework. To predict adverse drug-drug interaction (ADDI), the method MADRL implemented a discriminative learning model.
Recent approaches based on deep learning neural architecture have identified synergistic drug combinations for COVID-19. ComboNet predicts synergistic drug combinations for COVID-19.
The above-mentioned methods, though inspiring, do not incorporate chemical structure-derived features in the deep learning process. Recent advances like SSI-DDI and GMPNN-CS use Graphical Neural Networks (GNNs) using direct molecular structure information.
Graph neural networks in DGNN-DDI
GNN-based precursors to DGNN-DDI use different deep learning techniques, like graph attention layers (GAT) and message-passing neural networks (MPNN). These methods use the molecular graph representations of the drugs for determining the DDIs.
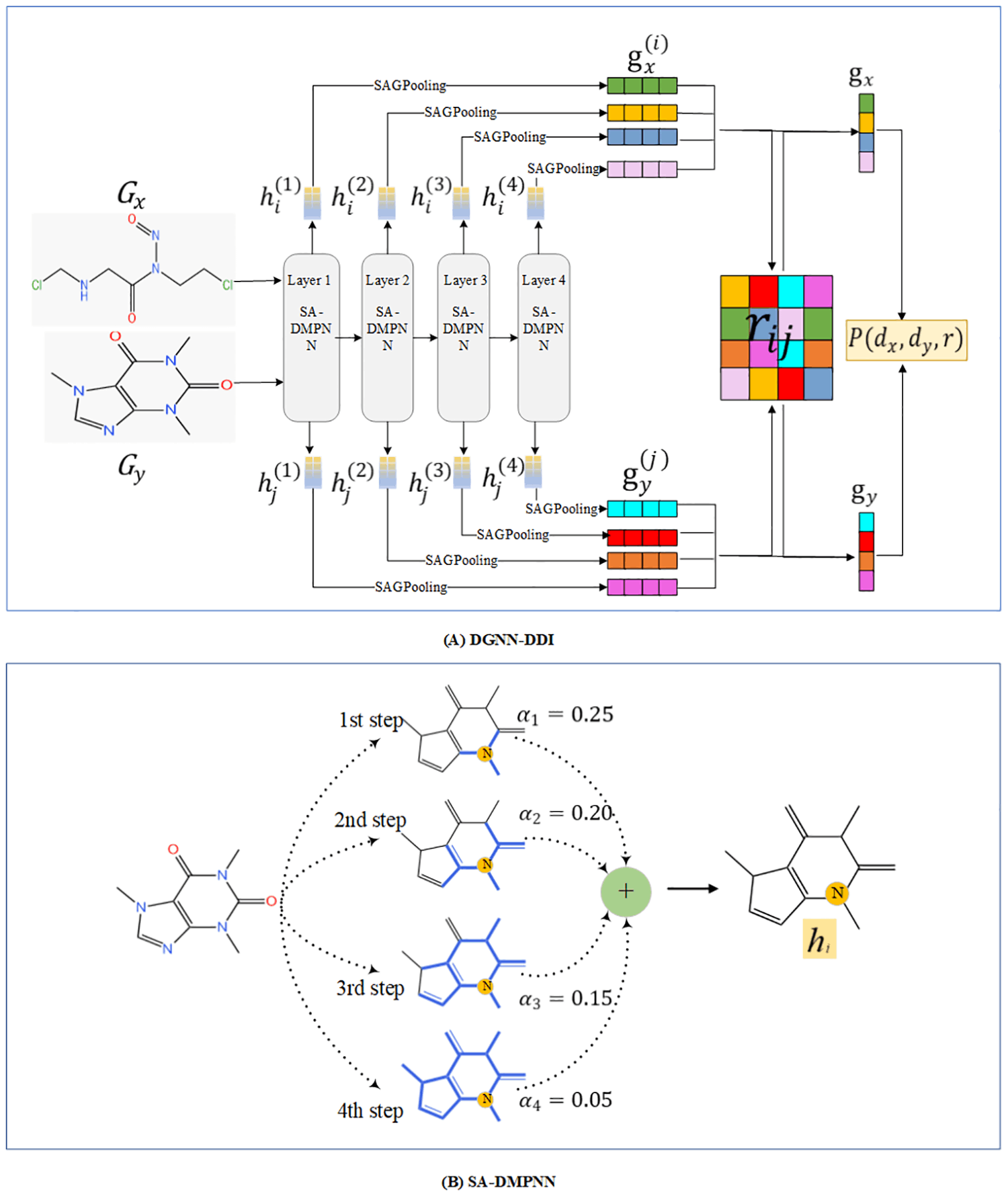
The authors propose a novel DDI prediction method called DGNN-DDI. This is a dual graph neural network-based DDI prediction tool, hence the nomenclature. The method takes into account the drug substructures as well as the interactions between the chemical substructures. Feature extraction for the method involves the following two major steps:
- In order to extract molecular substructure-related features, a directed message-passing neural network (DMPNN) is constructed. This DMPNN is endowed with a substructure attention mechanism (SA-DMPNN).
- Next, a co-attention mechanism, as in Lu JS et al., is introduced to determine the importance weights for the interaction scores between the two drug substructures.
DGNN-DDI’s approach
The model input is a DDI tuple (dx, dy, r), where dx and dy, both represented as SMILE strings, are the drugs, and r is a type of interaction. RDkit is used to preprocess the SMILE strings into graphs. So, a drug is represented by a graph G = (V, E), where V is the set of nodes, and E is the set of edges. Each node and edge has corresponding feature vectors.
For the graph G = (V, E), a GNN maps the graph to a d-dimensional vector hG in two phases: a message passing phase and a readout phase.
- Message-passing phase updates node-level features combining neighboring nodal features.
- The readout phase gives out a graph-level feature vector combining the node-level features. This output vector is used for predicting the label for the graph.
The following figure illustrates the string-to-graph conversion:
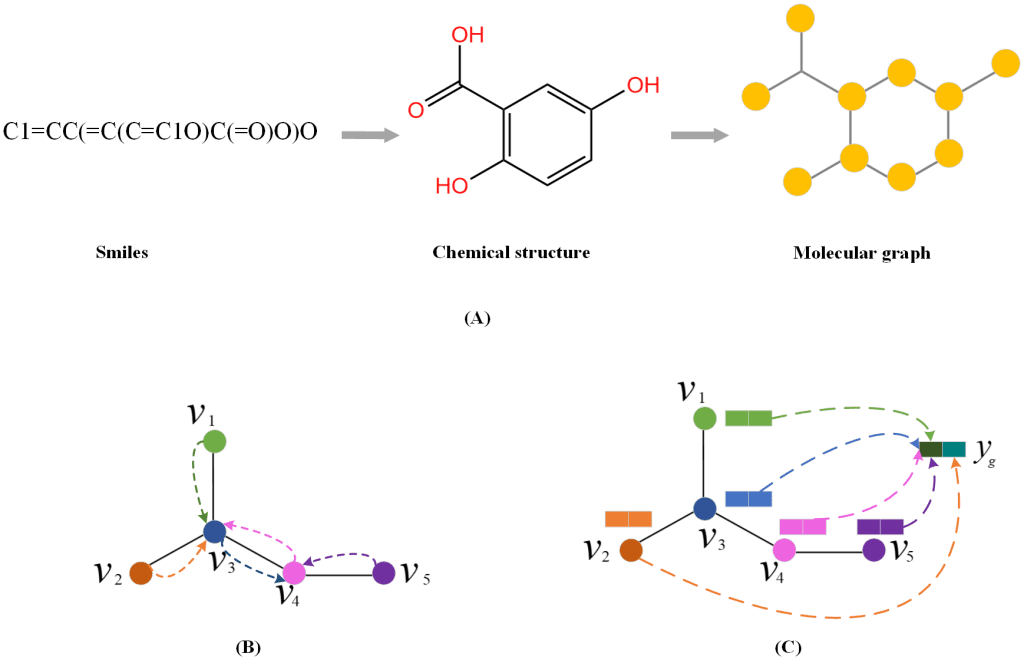
Image source:https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1010812
The following figure illustrates the major steps in the DGNN_DDI process:
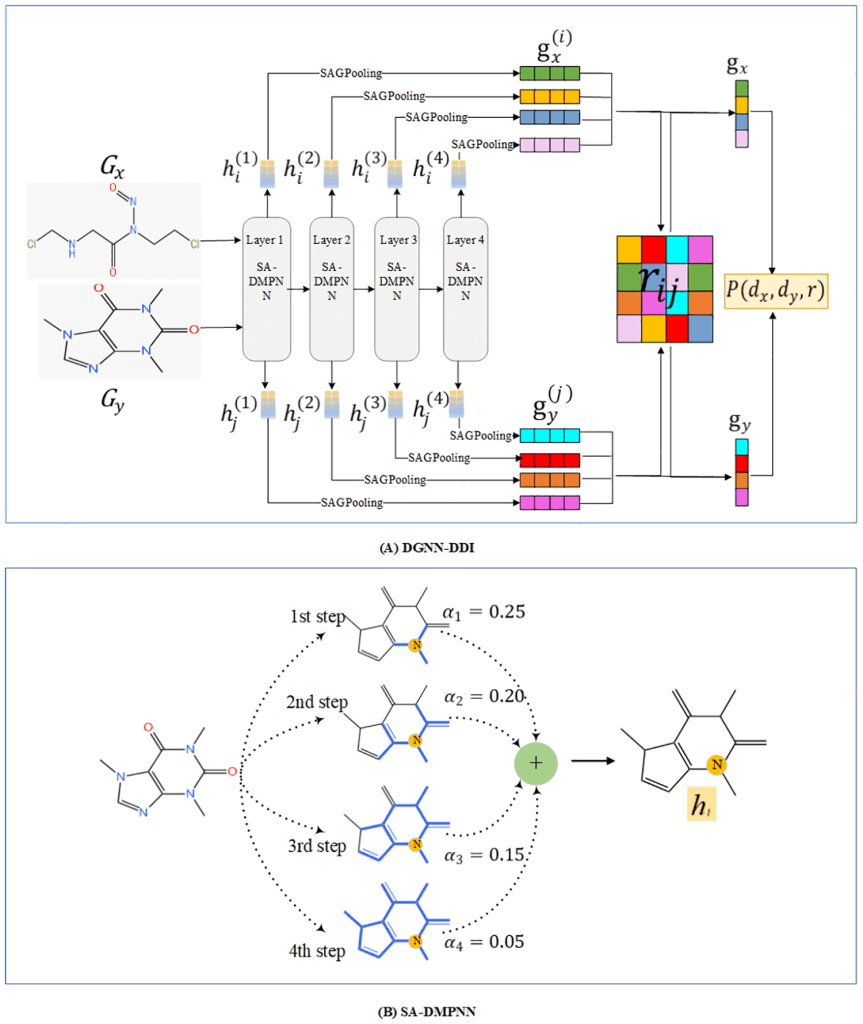
Image source: https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1010812
Novelty and scope
DGNN-DDI incorporates the molecular substructure drug features. DGNN-DDI also emphasizes and incorporates the interaction information between chemical substructures. Together combined, they yield better DDI predictions. The method has also been applied to real-world datasets to predict anti-COVID drug combinations.
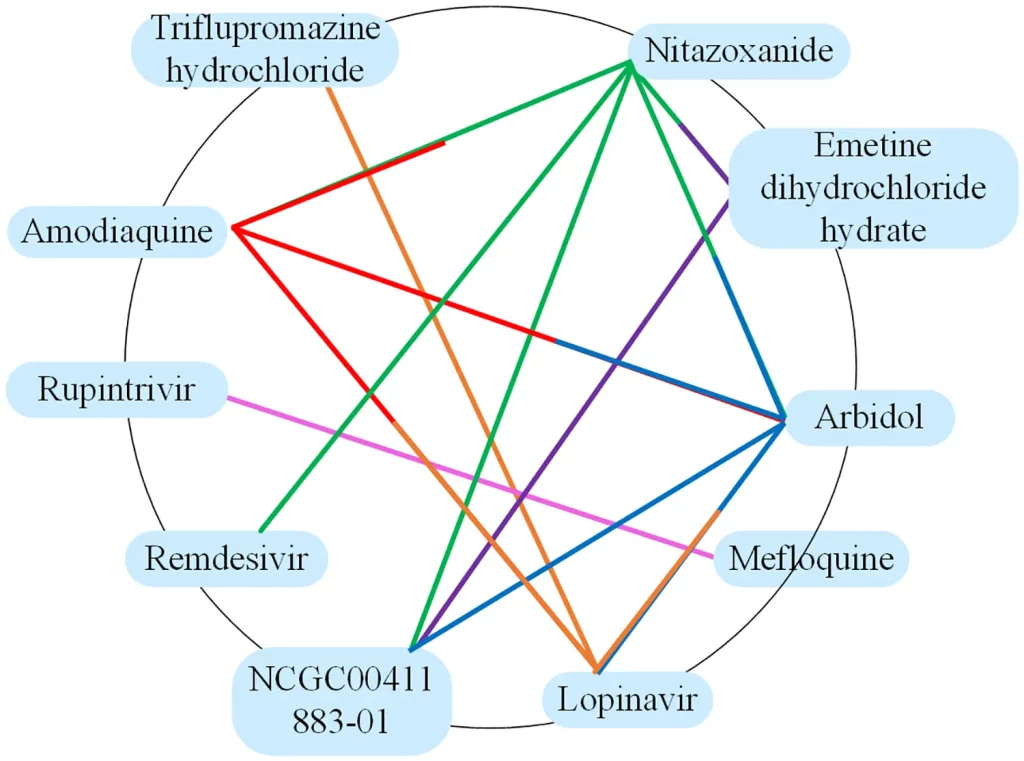
Image source: https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1010812
Outcome
- Comparative statistical analysis with cutting-edge approaches on the DrugBank dataset clearly shows that DGNN-DDI produces better performances as measured in various tests.
- The ablation study performed establishes the necessity of both the major components, SA-DMPNN and Multi-GNN, for better DDI predictions.
- DGNN-DDI is capable of detecting key substructures for DDIs.
Conclusion
In this study, a novel method incorporating molecular structure-based deep learning is developed for predicting DDIs. The co-attention mechanism, coupled with the substructure mechanism, yields better performance for DGNN-DDI in predicting DDIs as compared to other state-of-the-art methods available. DGNN-DDI has also delivered for combination drug therapy at the crucial juncture of the ongoing pandemic driven by COVID-19. The method clearly is a star addition to the bag of tools for DDI prediction, given its efficacy and better performance.
Article Source: Reference Paper
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Banhita is a consulting scientific writing intern at CBIRT. She's a mathematician turned bioinformatician. She has gained valuable experience in this field of bioinformatics while working at esteemed institutions like KTH, Sweden, and NCBS, Bangalore. Banhita holds a Master's degree in Mathematics from the prestigious IIT Madras, as well as the University of Western Ontario in Canada. She's is deeply passionate about scientific writing, making her an invaluable asset to any research team.





 prediction approach "DGNN-DDI" uses a dual graph neural network (GNN) to learn drug molecular features.){kind=link}