
Researchers from The Ohio State University utilized deep-learning classification and hierarchical stable-clustering approaches to predict cell types from single-cell mass cytometry data. The DGCyTOF approach could facilitate the analysis of single-cell RNASeq data and other omics data.
Single-cell mass cytometry, also called cytometry by time of flight (CyTOF), is a powerful high-throughput technology that permits the analysis of up to 50 protein markers for every cell to quantify and classify single cells.
The traditional manual gating used to identify new cell populaces has been inefficient, ineffective, unreliable, and hard to utilize, and no algorithms to identify both calibration and new cell populaces have been well established. Deep learning with graphic cluster (DGCyTOF) visualization has been created as a new integrated embedding visualization strategy for distinguishing canonical and new cell types.
The Working of DGCyTOF
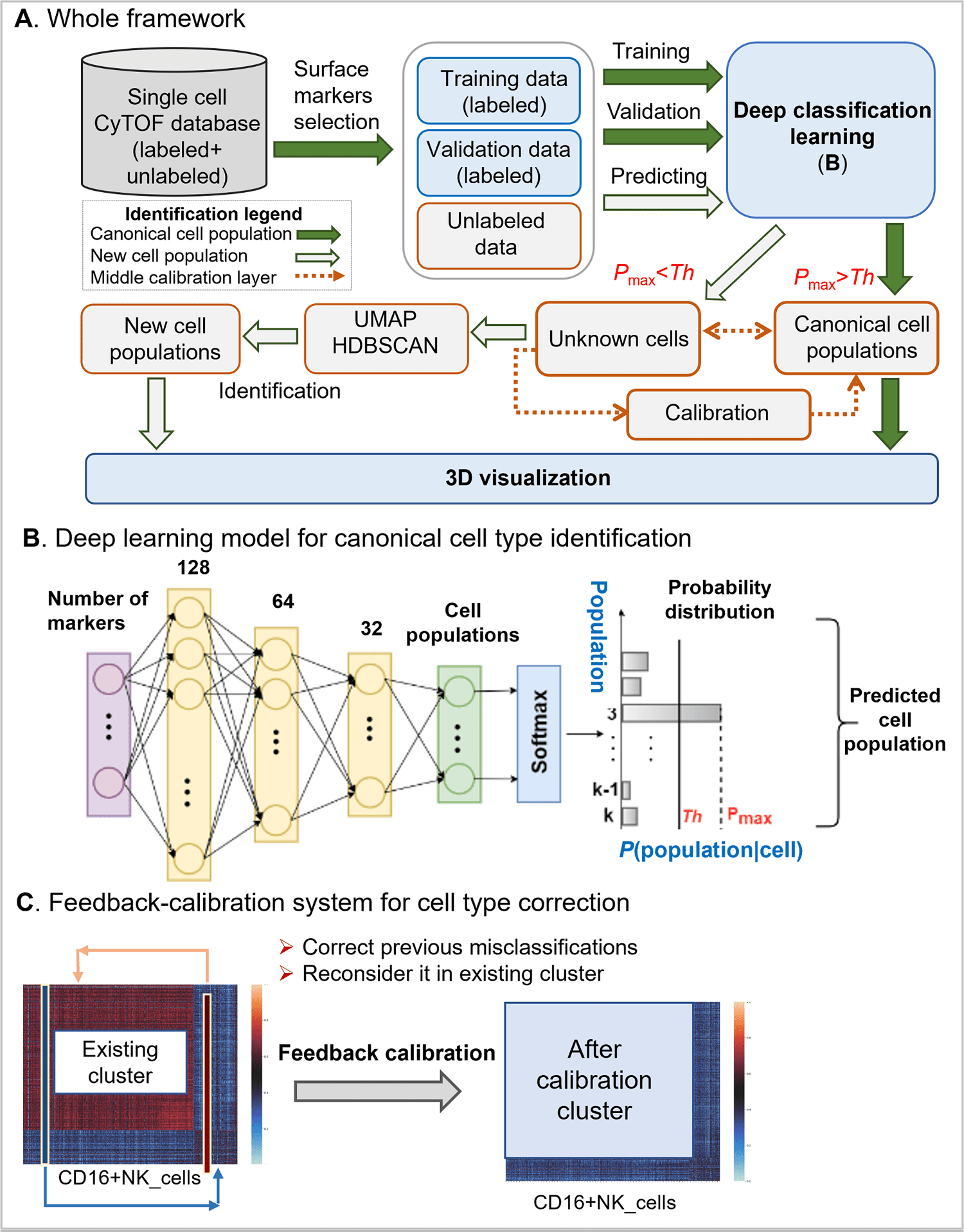
The DGCyTOF consolidates deep learning classification and hierarchical stable-clustering strategies to sequentially construct a tri-layer construct to identify known cell types and discover new cell types.
Image Source: DGCyTOF: Deep learning with graphic cluster visualization to predict cell types of single-cell mass cytometry data
To begin with, deep classification learning is built to distinguish calibration cell populaces from all cells by softmax classification assignment under a probability threshold, and graph embedded clustering is then used to determine new cell populaces sequentially.
In the center point of the two-layer, cell labels are adjusted automatically among new and unknown cell populaces utilizing a feedback loop using an iteration-calibration system framework to decrease the rate of error in the identification of cell types, and a 3D visualization platform is at last developed to show the cell clusters with all cell-populace types annotated.
Accuracy Comparison of Benchmark CyTOF Databases
By using two benchmark CyTOF information bases consisting of up to 43 million cells, the scientists compared the accuracy and speed in the identification of cell types among DGCyTOF, DeepCyTOF, and other different technologies involving dimension reduction with clustering, Principal Component Analysis (PCA), Factor Analysis (FA), Independent Component Analysis (ICA), Isometric Feature Mapping (Isomap), t-distributed Stochastic Neighbor Embedding (t-SNE), and Uniform Manifold Approximation and Projection (UMAP) with k-means clustering and Gaussian mixture clustering.
They noticed that the DGCyTOF addressed a robust and complete learning framework with high precision, speed, and visualization by eight estimation models.
The analysis of DGCyTOF with t-SNE and UMAP visualization in accuracy exhibited its around 35% prevalence in the prediction of cell types. Likewise, the visualization of cell populace distribution was more intuitive in the 3D visualization in DGCyTOF than in t-SNE and UMAP visualizations, respectively.
The DGCyTOF model can automatically assign known names to single cells with high accuracy by utilizing deep learning classification assembly with conventional graph clustering and dimension reduction strategies.
As directed by a calibration system, the model looks for optimal accuracy balance among calibration cell populaces and unknown cell types, yielding a complete and robust learning framework that is profoundly accurate in identifying cell populaces contrasted with results utilizing different strategies in the analysis of single-cell CyTOF information.
The utilization of the DGCyTOF strategy to identify cell populaces could be extended to the analysis of single-cell RNASeq information and other omics data.
Cell Type Identification by DGCyTOF in Two Datasets
The scientists utilized DGCyTOF to automatically identify known and new cell populaces in two benchmark CyTOF data sets resulting in F-scores of 0.9921 (CyTOF1) and 0.9992 (CyTOF2) for the identification of labeled cells and cell populaces.
The results of graphical clustering depicted unlabeled cells not belonging to known populaces were parted into five clusters in the CyTOF1 dataset. In contrast, the results of spectral clustering depicted new clusters of cell populaces among unknown cells. Finally, by using the DGCyTOF platform, the final cell populaces were displayed in a 3D space which included new subtypes and calibration (known) cell types.
Externally Validating the DGCyTOF Model in the Identification of Known Cell Types
External validation was utilized to assess the overfitting phenomenon in the deep learning algorithm. The scientists randomly separated all labeled samples equally into a training set and a validation set for CyTOF1 and CyTOF2.
They utilized weight regularization to diminish the overfitting of the Deep Learning (DL) model. The hyperparameter tuning of the DGCyTOF model was done by the utilization of a regularization l1 normalization to penalize weights sparse to 0, and they chose the top 5% parameters.
Then again, the researchers designed the dropout and dense strategy for parameter selection to keep neural networks from overfitting.
The DeepCyTOF platform chose parameters automatically by its design. The confusion matrix permitted the researchers to visualize the performance of the supervised algorithm, wherein each row of the matrix represented the instances in an actual class, and every column addressed the instances in a predicted class or vice versa.
The researchers applied a ROC metric to assess classifier output quality by utilizing 4-fold cross-validation between the true and prediction labels.
The results showed the average performance in the two testing datasets, with 71.9 to 99.4% accuracy and average ROC area of 0.94 to 1.00 in CyTOF1 and 96.5 to 99.8% precision and average ROC area of 1.00 in CyTOF2.
They then made a comparison of all prediction results for combination methods with true labels, utilizing four evaluation criteria to make a comparison of the performance of the DGCyTOF and DeepCyTOF techniques in classifying marked information in the CyTOF1 and CyTOF2 data sets:
- F-score (harmonic mean of precision and recall);
- ARI (adjusted Rand index, a measure of similarity between two clusters);
- FMI (Fowlkes-Mallows scores); and
- V-measure (consonant mean among homogeneity and completeness).
Conclusion: The High Potential of DGCyTOF
Recent advancements and innovations in mass cytometry (CyTOF) have profoundly altered the destiny of single-cell proteomics by permitting a more precise comprehension of complex biological frameworks and the identification of novel cellular subsets.
Mass cytometry permits analyses of cells in suspension-like blood yet is additionally extended for the analyses of tissue segments. The new calculational technology needed in managing such big data to describe the complex cellular sample types, where rare cell populaces with fundamental biological functions would somehow be missed.
Interestingly, the researchers propose the DGCyTOF method by incorporating the upsides of both the classifier and cluster strategies in the identification of known and new cell types as indicated by relative protein abundances from cytometry information.
The DGCyTOF strategy permits automatic and exceptionally accurate assignment of known labels to single cells utilizing deep learning, identifies new cell populaces using an original novel graphical clustering technique, and utilizes the guidance of a calibration system to accomplish an optimum balance of accuracy.
Directed by a feedback calibration system, the model looks for an optimal accuracy balance among calibration cell populaces and unknown cell types, yielding a complete and robust learning framework that is exceptionally accurate in the identification of cell populaces contrasted with results involving different strategies in the analysis of single-cell CyTOF information.
The researchers believe that the novel DGCyTOF will distinguish the cells into functionally distinct groups and types, thereby allowing point-by-point detailed analysis of cell heterogeneity not for calibration cell types but for new rare cell types.
The DGCyTOF holds incredible potential to reveal the tissue and immune system’s cellular variation patterns and functionality by these inferring cell types. The use of the DGCyTOF strategy to identify cell populaces could be extended to the analyses of single-cell RNASeq information and other omics data.
Paper Source: Cheng L, Karkhanis P, Gokbag B, Liu Y, Li L (2022) DGCyTOF: Deep learning with graphic cluster visualization to predict cell types of single cell mass cytometry data. PLoS Comput Biol 18(4): e1008885. https://doi.org/10.1371/journal.pcbi.1008885
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Tanveen Kaur is a consulting intern at CBIRT, currently, she's pursuing post-graduation in Biotechnology from Shoolini University, Himachal Pradesh. Her interests primarily lay in researching the new advancements in the world of biotechnology and bioinformatics, having a dream of being one of the best researchers.











{kind=link}