
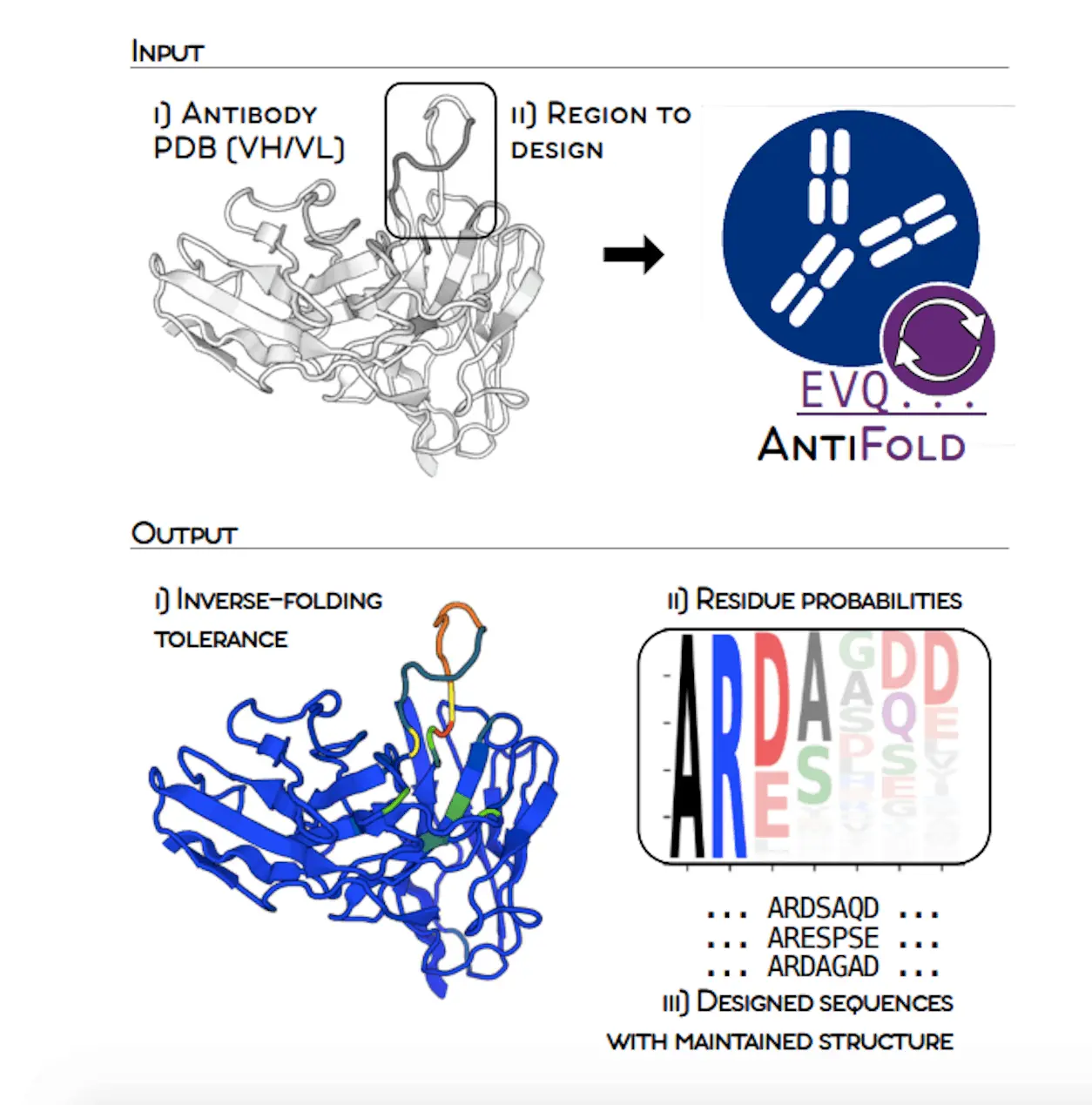
Scientists from the University of Oxford along with the Technical University of Denmark have introduced AntiFold: a specialized model for antibody design that will help in improving sequence recovery in the most important regions and, at the same time, will aid in the zero-shot affinity prediction. It is the antibody structure that is the input, and by its means, we can know the mutation tolerance without changing the backbone structure. The structurally stable sequences are designed to maintain the structural integrity, thus helping the antibodies to be targeted effectively, which in turn would lead to better therapeutics.
Antibodies are a type of therapeutic that can be used to cure many diseases. However, they can be difficult to design because they require the optimization of several properties such as efficacy, manufacturability, and safety. Machine learning methods have turned out to be the tools that are very valuable in this process, thereby facilitating the antibody development in such a way that it addresses the issues related to immunogenicity and aggregation and at the same time it increases the desirable properties like binding affinity. However, the issue is picking changes that preserve the antibody structure while increasing its characteristics.
To solve this issue, Researchers present AntiFold, a specialty of the antibodies-specific inverse folding model, which is the result of ESM-IF1. Unlike the other tools, AntiFold greatly improves the sequence recovery in the hypervariable CDR regions and provides the ability to predict the affinity without any samples. The method assesses the tolerance of mutations without the need to change the backbone structure and the probability of each amino acid position. Users can, if they want, set the regions of the sequences and tune the temperature parameter to regulate the diversity of the sequences. AntiFold designer sequences have a high structural similarity to the original structure upon re-folding, which is very important.
AntiFold, along with other property prediction tools, gives a strong weapon for directing the antibody optimization process by reducing the range of options for experimental testing. The combined way of working could make the development process of antibodies simpler and more efficient. Thus, the discovery of new and better therapies could be increased.
Data Collection
The advantage that AntiFold has is its ESM-IF1 pre-training on the over 12 million structures, which enables it to be trained further on both solved and predicted antibody structures. For a fair comparison with AbMPNN, the researchers utilized the same dataset: Filling the gap of 147,458 sequences from the Observed Antibody Space (OAS) paired database, the ABodyBuilder2 model solved 2,074 cases from the Structural Antibody Database (SAbDab). The data was divided into training, validation, and testing sets using a 90% split of the sequence identity on the concatenated CDR sequence based on the 80/10/10 cutoff. However, since they could not directly compare it with IgMPNN because of the unavailability of the model weights and training data, AntiFold’s performance is still remarkable in this context.
Training of AntiFold
In the process of fine-tuning, Researchers came up with different methods to improve the performance in the validation set, especially in the CDRH3 loop, which is the amino acids recovery, i.e., essential for antigen binding. These strategies included:
- The span-masking scheme, which showed a 36.4% improvement.
- Additional masking of random residues (53.2%).
- Weighting masking towards CDR residues with a 3:1 weight (53.3%)
- Rate-of-decay per layer (54.4%)
- Predicted structures from OAS (58.4%)
These efficiencies were included in the AntiFold model, which was finally developed. To clarify, the fact that AntiFold was trained without the ESM-IF1 effectiveness was the reason the CDRH3 AAR level was 31.5% on the validation set is one of the major reasons for the high accuracy of ESM-IF1, as it takes advantage of the huge ESM1 pre-training dataset.
Results
- Improved Amino Acid Recovery on antibody sequences by Fine-tuning
AntiFold displayed a remarkable improvement in amino acid recovery (AAR) on the experimental structure test set as compared to the original ESM-IF1 model, especially noticeable in the CDRH3 region (60% vs 43% AAR, p < 0.005). Furthermore, AntiFold was better in most of the CDR and framework (FR) regions than AbMPNN. Importantly, AntiFold was steadily performing well on both solved and predicted structures as opposed to AbMPNN (AAR -2.7%), which was slightly losing its performance on experimental structures.
- High Structural Agreement of Predicted Sequences:
To check the structural reliability of sequences proposed by AntiFold, researchers sampled and refolded CDR sequences from a dataset of 56 high-quality antibody structures that were determined using X-ray crystallography (resolution < 2.5 Å). Using a residue sampling temperature of 0.20, they acquired 20 sequences for each antibody.
AntiFold kept the original molecular backbone in its structure with high structural fidelity; the mean CDR region root-mean-square deviation (RMSD) of 0.95 was the result of that. On the other hand, AbMPNN, ESM-IF1, and ProteinMPNN had the highest RMSD values (0.98, 1.01, and 1.03, respectively), while the native RMSD was 0.63. This proves that AntiFold has the best skill to keep the backbone structure which means it could be a good tool to use for designing antibodies that will be easier to build and will have a very small disturbance.
- Prediction of Antibody-Antigen Binding Affinity:
AntiFold proved to be the best in the forecasting of antibody-antigen binding affinity using a deep mutational scan of an anti-lysozyme antibody among the inverse folding models. This was revealed by its remarkably high Spearman’s rank correlation coefficient of 0.418, versus ESM-IF1 (0.334), AbMPNN (0.322), ProteinMPNN (0.301), and ESM-2 (0.264), assessed by Mann-Whitney one-tailed U tests. The effectiveness of AntiFold increased even more if the antigen chain(s) were included, especially in CDR regions, in particular the CDR2 (Sr 42.7 to 47.3%, p < 0.005) and CDR3 (Sr 29.8 to 32.0%, p < 0.005), but not for framework residues. At the same time, other models showed that their performance was reduced when the antigen chains were taken into account.
To deepen the understanding of AntiFold’s ability to predict the binding affinity and, hence, guide the antibody design, researchers tested the model on 124 variants of 7 antibodies that were generated using protein language model-guided affinity maturation experiments. Upon the variation score normalization and the subsequent evaluation of the variation ranking against the experimentally measured variants, AntiFold was able to significantly better the separation of the lower, maintained, and improved binding affinity groups. The optimized versions were graded with a median rank score of 80%, while ProteinMPNN had 73%, ESM-IF1 had 57%, and AbMPNN 55%, which shows the goodness of AntiFold in the guiding of antibody design efforts.
Conclusion
In conclusion, AntiFold, which was refined from the ESM-IF1 model, reached the best performance in antibody sequence recovery and refolding. Pre-trained weights are the main contributors to the performance of a model, and when predicted structures are added and weighting masking toward CDR residues, it enhances the model’s performance even more. The probability of AntiFold’s inverse folding corresponds very well with the antibody-antigen binding affinity, particularly when the antigen information is set as a parameter. It successfully uncovers the regionally structurally limited, high-fitness zones of the mutational landscape; hence, the binding is preserved.
Since the scientific community is still investigating the possibilities of computational models in protein engineering, AntiFold has emerged as a potential solution to the problems of antibody optimization. Thus, it will lead to further discoveries of the new field in the future.
Article Source: Reference Paper | AntiFold is freely available under the BSD 3-Clause as a web server and pip installable package at GitHub.
Important Note: arXiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Anshika is a consulting scientific writing intern at CBIRT with a strong passion for drug discovery and design. Currently pursuing a BTech in Biotechnology, she endeavors to unite her proficiency in technology with her biological aspirations. Anshika is deeply interested in structural bioinformatics and computational biology. She is committed to simplifying complex scientific concepts, ensuring they are understandable to a wide range of audiences through her writing.











{kind=link}