
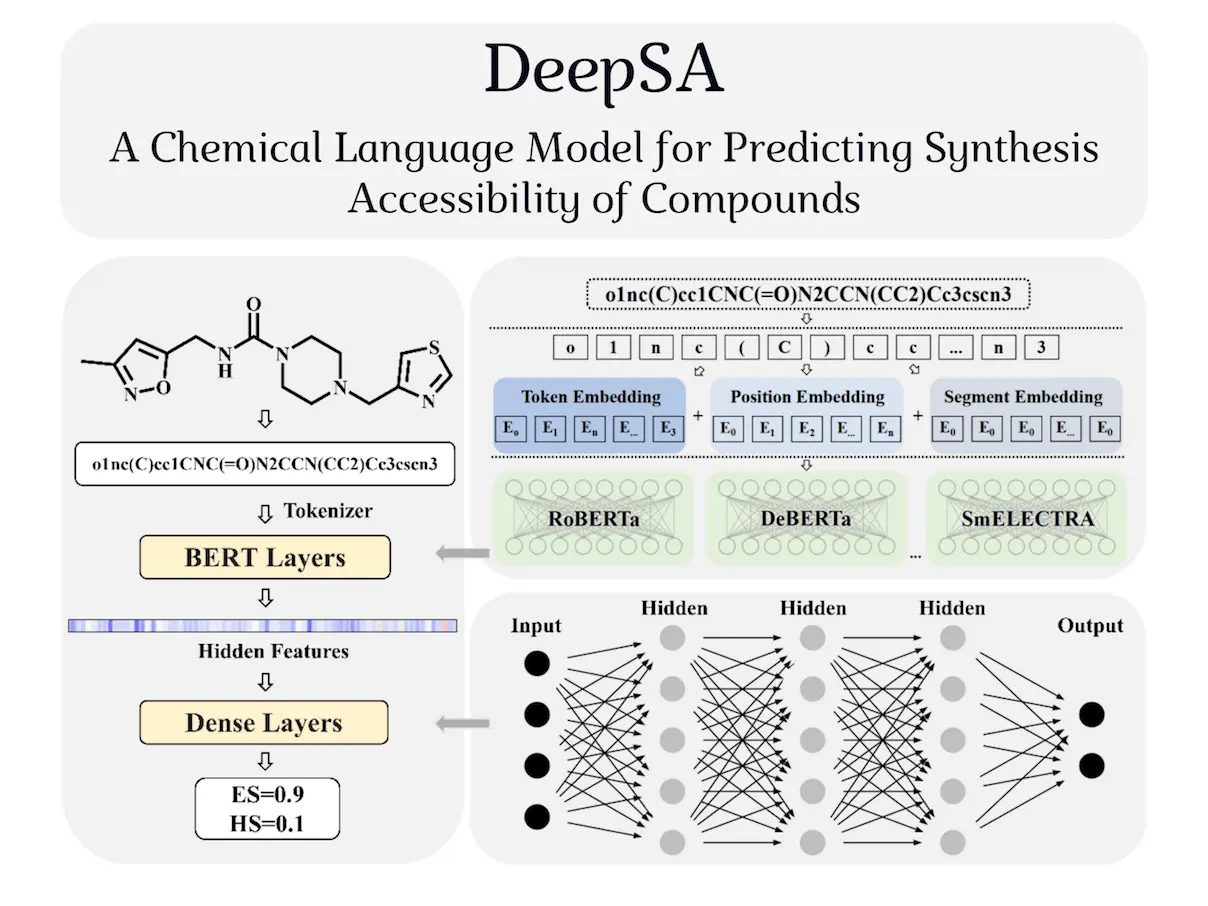
As computationally generated molecules become more popular in the fields of drug development and biotechnology, ease of synthesis becomes a common problem. Often, the molecules generated by machine learning models may be challenging to synthesize and may require many steps, making it too expensive for production. A new deep learning model DeepSA, developed by researchers from ShanghaiTech University, China, seeks to solve this problem by identifying the synthetic accessibility of various compounds without the need for human input.
With new developments in artificial intelligence, the use of computational tools to generate novel molecules has become much more widespread. However, a few problems arise with computationally derived molecules, namely how easy they are to synthesize: a characteristic known as a compound’s synthetic accessibility. Computational capacity to generate new molecules is mostly utilized in the field of computer-aided drug design and, more specifically, AI-aided drug design. This drastically reduces the time needed to formulate new compounds and reduces expenses significantly. CADD is performed using many methods, one of which is fragment-based drug design. Using a specific structure as a target, this method involves the performance of a virtual screening on a library of molecular fragments to procure ligand fragments, after which several rounds of optimization and modification are performed according to the required parameters to obtain the desired compound. However, some compounds achieved through this process can be challenging to synthesize, making them too costly for industrial production.
Machine learning can be an appropriate solution to such a problem, as with appropriate training, it should be possible for a deep learning model to identify which molecules are viable in terms of synthesizability. Tools for this purpose have been created using various methods, such as SAscore, SCScore, and RAscore, which utilize historical data from multiple datasets to score compounds’ synthetic accessibility on a scale of 1 to 10.
A new model has been proposed to evaluate this characteristic using a chemical language model. DeepSA has greater accuracy than other models when differentiating compounds that are easy or difficult to synthesize. Compared to other models that perform the same function, DeepSA’s ability to identify the synthetic accessibility of compounds was found to be better and more aligned with the results from lab testing.
The same datasets used for the training of prior models were also used to train DeepSA in order to ensure that any superiority in performance was due to the methodology rather than a difference in training sets. More than 800,000 different molecules were present in the training sets, and their synthetic accessibility was evaluated using Retro*, a multi-step retrosynthetic planning algorithm. Retro* is a neural-based algorithm capable of finding simple synthetic routes for target compounds. The SMILES for the molecule were used as input for the algorithm in order to find the final number of steps required to synthesize it. If the number of steps was below 10, it was classified as easy to synthesize.
After this, another 650,000 molecules were utilized using different SMILES representations for the addition of more complicated sampling operations to the datset provided. The test sets used to evaluate the model performance independently were drawn from three independent studies. Statistical indicators like accuracy, precision, and F-score were used to evaluate the models’ performances.
DeepSA comprises three different modules – the first being the data processing module, the second being the feature embedding module, and the last being the decoder module. The original SMILES given in the dataset were converted to canonical SMILES, after which the dataset was expanded further through the introduction of alternative SMILES formats. A tokenizer was used to encode compound structures, and basic atomic structures used in input were treated as separate “words” to make coherent predictions. Network architectures were used from natural language models and chemical language models to arrive at the final product. The models’ architectures were optimized using the modified dataset so it would be suitable for the task of determining synthetic accessibility.
For further validation, the model’s performance for 18 compounds was assessed and compared to synthetic pathways previously published. These compounds were not present in the training sets. The model was successful in predicting the synthetic accessibility of all compounds. The model has also been deployed online for researchers, allowing for greater accessibility to the software.
The development of machine learning techniques for natural language processing has resulted in the creation of various natural language models. These models are capable of processing protein sequences the way they do natural languages. It has been noticed that SMILES sequences, which represent various compounds, are similar in structure to natural language, allowing these frameworks to be used with minor modifications. Having simple characters that combine to create a large vocabulary, as well as creating complex sequences through simple rules, are things that SMILES sequences and natural languages have in common with each other.
Conclusion
Similar training strategies to those used for natural language models can thus be utilized for deciphering chemical language as well. With extensive training, the model can be made to identify patterns and trends in these languages and use these to generate conclusions about new inputs. While this means the capabilities of the model are restrained by the diversity of data that they are trained on, this allows for great consistency and accuracy in the results obtained in much shorter periods of time. This makes them greatly useful for research purposes, where budgets and resources are often thin, and can facilitate the widespread adoption of computer-aided drug design.
Article source: Reference Paper | DeepSA is available online at the Web Server | DeepSA code is available at GitHub
Learn More:




Sonal Keni is a consulting scientific writing intern at CBIRT. She is pursuing a BTech in Biotechnology from the Manipal Institute of Technology. Her academic journey has been driven by a profound fascination for the intricate world of biology, and she is particularly drawn to computational biology and oncology. She also enjoys reading and painting in her free time.











{kind=link}