
Chemical language models (CLMs) can be used to construct molecules with desired features. Textual representations of novel chemical structures, such as simplified molecular input line entry system (SMILES) strings, are generated using CLMs. The ability of generative models to learn a training distribution of molecules determines their usefulness for designing new functional substances. Through the computational development of phosphoinositide 3-kinase gamma (PI3Kγ) ligands, this study demonstrates that hybrid CLMs can benefit from the bioactivity information supplied for the training molecules.
Requirement for Chemical Language Models (CLMs)
While the ultra-large and potentially unlimited size of virtual compound libraries give us access to previously unexplored chemical space, there have been questions about the practicality of screening billions of molecules with a possible massive risk for false positives. Researchers have used generative deep learning models to address some of these issues to get smaller, focused virtual compound libraries and create compounds on demand through de novo (from scratch) design.
Chemical language models are neural network-based systems that process string representations of molecules (for example, simplified molecular input line entry system (SMILES) strings) and have previously been utilized to construct specialized virtual chemical libraries successfully. In this study, a data-driven molecular design process was established that used structural and bioactivity information from known ligands to produce custom molecules by learning from a textual description of molecules. Two CLMs were pre-trained on a vast collection of patented compound structures, each with its pretraining technique. The CLMs were fine-tuned by inhibitors of phosphoinositide 3-kinase gamma (PI3Kγ), a therapeutic target for anticancer, anti-inflammatory, and immunomodulatory drugs.
Why Phosphoinositide 3-Kinase Gamma?
Phosphoinositide 3-kinase gamma is a promising therapeutic target for treating advanced solid tumors and autoimmune diseases. Another reason for choosing phosphoinositide 3-kinase gamma was based on the availability of data in the DTC database.
CLM for De Novo Production of a Targeted Chemical Library for PI3Kγ
For the de novo production of a targeted virtual chemical library for PI3Kγ, a CLM based on a long short-term memory (LSTM) neural network with SMILES strings as input was constructed. The CLM was trained specifically using an autoregressive technique, which is the process of continuously predicting the next character in a SMILES string given all of the previous characters in the string.
Chemical language models generate new molecules by stretching strings from a “start” character until the string reaches a predetermined maximum length or until the “stop” character is sampled. The probability distribution that the CLM learned during training is used to insert string characters repeatedly. According to the probabilities learned by the CLM, the more likely a specific character is at a given step, the more frequently it will be sampled, and vice versa. In this study, ‘nucleus sampling’ was adopted, which narrowed down the probabilities learned by the CLM and stopped it from selecting unlikely SMILES characters. A probability criterion of 0.85 was employed to construct a PI3Kγ focused library.
Pretraining CLM for Prediction of Bioactivity
Due to the availability of bioactivity data for fine-tuning molecules, a bioactivity prediction algorithm was trained to choose the best de novo designs. The study’s purpose was to investigate the capability of a SMILES string-based hybrid CLM to determine bioactivity. With a large amount of unlabeled data, two pretraining strategies were used for feature learning: Autoregressive pretraining (similar to the one used for generative CLM) and ELECTRA (Efficiently Learning an Encoder that Classifies Token Replacements Accurately) pretraining.
In the ELECTRA approach, the training data consisted of corrupted input SMILES strings that were created by arbitrarily replacing multiple characters with other SMILES language characters. The model was trained to recognize these. It was found that the ELECTRA method produced better results than the standard approach when identifying the most active molecules and limiting the falsely labeled ones. For the same true positive rate of 71.3%, the model pre-trained using ELECTRA had a false positive rate of 10.0%, compared to the 46.7% of the standard CLM model at threshold 0.4. The majority voting deep ensemble (which integrated predictions from several different models) increased confidence levels, resulting in a fall in the number of projected “highly active” molecules.
Testing Compounds Produced by the CLM
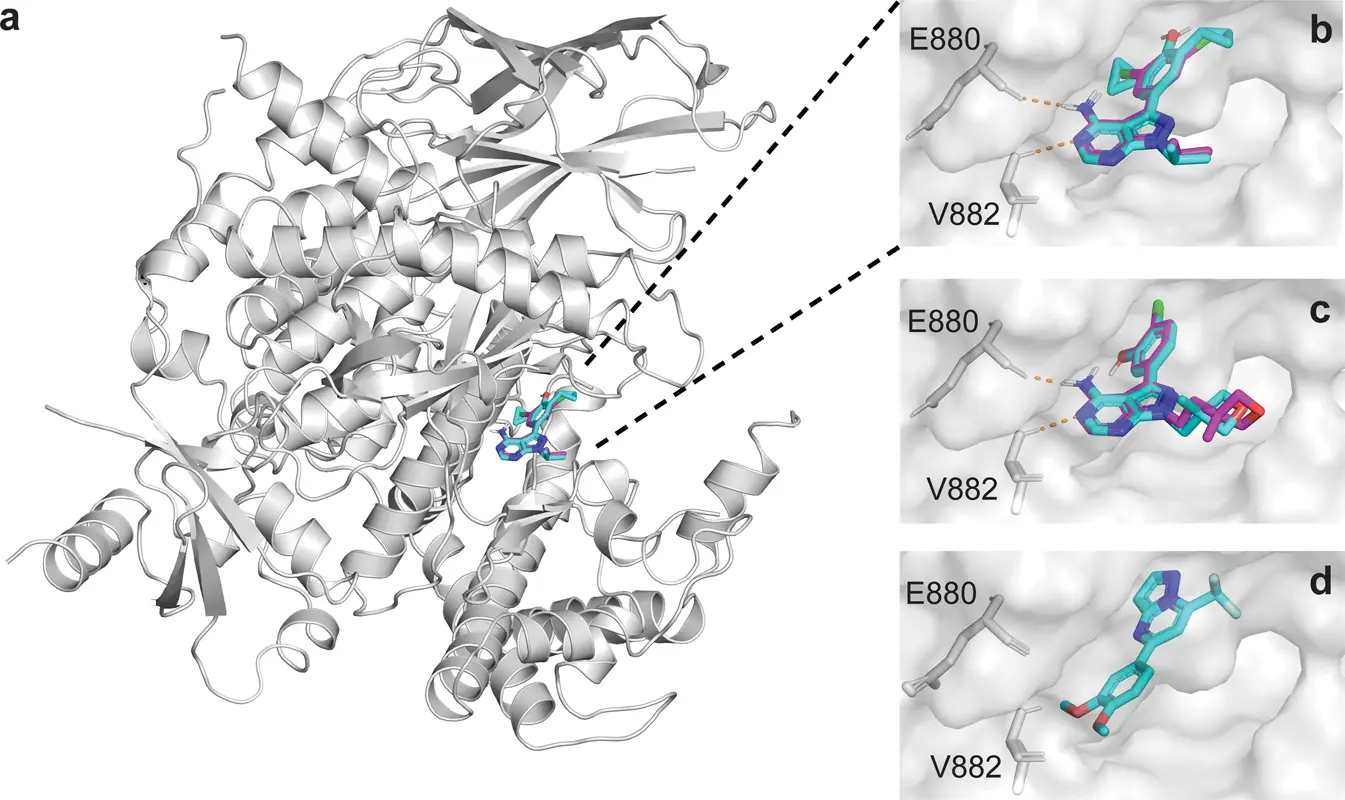
Some of the compounds produced by the CLM were evaluated in vitro for PI3Kγ binding as evidence of concept. Test compounds from improved virtual chemical libraries were selected to be acquired commercially rather than through the synthesis of de novo designs to maximize efficiency in terms of time and resources. There were 16 molecules available, with compound 1 being the highest ranked with 80/100 votes. The lower-ranked molecules were unable to inhibit PI3Kγ in the assay. Compound 1 consisted of a “scaffold hop” (modified core structure) from well-known inhibitors. However, the presence of a de novo design with 99/100 votes shows generative molecular design’s potential to explore the chemical universe beyond commercially accessible chemicals.
After receiving 99/100 votes, two of the top-ranked computer-generated molecules (Compounds 17, 20) and their derivatives (Compounds 18, 19, 21, 22) were synthesized. Because the ELECTRA-CLM score alone was insufficient to differentiate between the molecules, the TIGER software separated them and categorized them according to their scaffolds. Following synthesis and purification, these were tested for direct PI3K binding, with compounds 18 and 22 proving to be the most effective. These two compounds were subsequently tested to verify their biological actions. They demonstrated that both compounds, at low nanomolar concentrations, suppress the activation of the PI3K effector kinase AKT under physiologically relevant circumstances in human tumor cells.
Conclusion
This research shows the adaptability of deep generative learning for hit and lead finding in the development of drugs. De novo drug design aims to make novel molecular scaffolds with desirable qualities while recommending subtle adjustments for lead optimization. The hybrid CLM classifier used structural and bioactivity information from fine-tuning molecules to create a virtual chemical library, expanding the methodological capacity for virtual screening. The findings strongly support using hybrid CLMs for activity-focused molecular design and virtual compound screening. Three de novo designs were tested successfully for bioactivity. Whether one de novo design is better than another depends on the task at hand, making any overall method evaluation difficult. Therefore, practical application is the best judge.
Article Source: Reference Paper
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Sejal is a consulting scientific writing intern at CBIRT. She is an undergraduate student of the Department of Biotechnology at the Indian Institute of Technology, Kharagpur. She is an avid reader, and her logical and analytical skills are an asset to any research organization.





{kind=link}