

Targeting nucleic acid macromolecules, particularly RNA, for structure-based drug design (SBDD) is gaining research attention as it resulted in several FDA-approved compounds targeting RNA molecules.
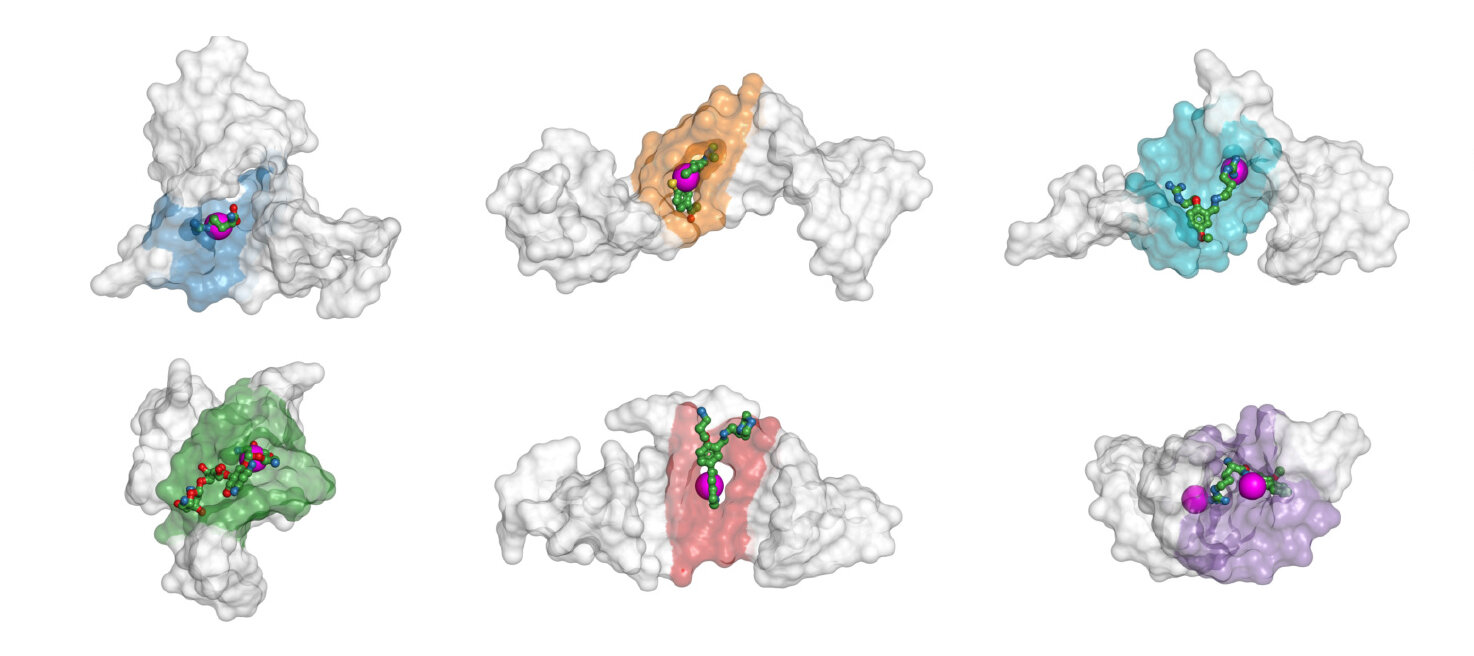
Image Source: https://www.skoltech.ru/en/2022/01/artificial-intelligence-predicts-rna-and-dna-binding-sites-to-speed-up-drug-discovery/
Scientists from Skoltech’s iMolecule group have created an artificial intelligence-driven approach, “BiteNetN,” to identify sites on the structures of DNA or RNA molecules where drug compounds may bind. The drug-binding site information will allow pharmaceutical firms to find novel therapeutic compounds – including antiviral agents – in a far more focused manner. The new method, which was published in Nucleic Acid Research: Genomics and Bioinformatics, claims to be more accurate than previous methods since it considers how a nucleic acid molecule’s shape impacts which binding sites are accessible.
Most drugs target proteins because pharmacologists have traditionally seen RNA as just a mediator between DNA and the functional proteins it encodes. As almost 85% of the genome is translated into RNAs, only a tiny percentage of those RNAs encode proteins. The remaining noncoding RNAs regulate specific genes or perform other functions by adopting different conformations. Because noncoding activities can also have a pathogenic component, RNA and perhaps DNA sequences are more widely recognized as therapeutic targets.
“Nucleic acids -; DNA and RNA -; can participate in signaling, for example, and we could target that or any other process they are involved in. This could be a promising strategy for undruggable protein targets, for example, disordered proteins or proteins that lack convenient binding sites. And then there’s also pathogenic RNA foreign to the body, for example in viruses, such as SARS-CoV-2 or HIV.”
Petr Popov, principal investigator of the study, Skoltech Assistant Professor
To find out the potential inhibitors of a drug target, pharmacologists need techniques for screening enormous libraries of chemical compounds to identify which interact with nucleic acids and where the specific binding sites are.
“We created this new solution by adapting our prior work with proteins,” Popov explained. “Nucleic acid three-dimensional structures are encoded as high-dimensional tensors. Once this is done, a computer vision algorithm ‘looks’ at the tensors and highlights the areas in the structure that it thinks could serve as binding sites. After the confirmation and the binding site have been detected, a more focused drug discovery campaign can be initiated. So our work is a small step toward rational drug discovery in contrast to the blind screening, which becomes less reliable with growing chemical libraries.”
There’s also a twist involving the form of the RNA and DNA molecules. They tend to twist and tangle into various shapes. The characteristics of the molecules are altered as a result of these so-called conformation modifications, including which binding sites are accessible. Conventional methods target nucleic acid sequences and ignore conformation, making them erroneous.
According to Igor Kozlovskii, the first author of the paper, “Most earlier methods only worked with RNA, and specifically, with a single chain. Ours works with DNA and with two or more chains. We can even see additional sites that arise when multiple molecules become entangled,”.
“A great example of what makes working with methods that ignore conformation problematic is the dominant type of HIV,” he went on. “It has an RNA region targeted by many agents. But even though the nucleic acid sequence is the same, when that molecule changes conformation, this is known to have an effect on which agents work or don’t. Our neural network predictions actually reproduce this effect, which means they are reliable.”
The new approach has an interesting application: it allows you to use the procedure “in reverse.” Rather than identifying binding sites on a prospective target, the algorithm may focus on a harmful agent, such as a tiny hormone molecule, which may be causing a condition and distract it.
“So we want to bind those small molecules with something. To do it, we need to reverse-engineer a short nucleic acid fragment, called aptamer, that would serve as a decoy for the hormone or other molecule of interest. Naturally, an aptamer must contain a binding site, and our solution can be applied to design aptamers with improved binding properties,” concluded Popov.
Story Source: Igor Kozlovskii et al., Structure-based deep learning for binding site detection in nucleic acid macromolecules, NAR Genomics and Bioinformatics (2021).
Story Source: Reference Paper | Reference Article | BiteNetN is publicly available on the Website
Learn More:



Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.











{kind=link}