
Deep Docking (DD) is artificial intelligence (AI) driven method that provides very economical yet highly reliable access to ultra-large docking. It was developed by Scientists at the University of British Columbia, Vancouver, to address the global lack of computational docking power and to enable structure-based screening of chemical libraries having billions of molecules without using extraordinary computational resources.
The rapid expansion of make-on-demand libraries to billions of synthesizable compounds has sparked the interest of the drug-discovery community because such ultra-large databases allow access to novel, unknown portions of the chemical universe. With chemical libraries containing 100 billion molecules on the horizon, traditional docking technologies will need to be modified.
This method uses small batches of explicitly docked compounds to iteratively train deep neural networks (DNNs) and use it to predict the ranking of the library’s remaining unprocessed compounds. Deep Docking can eliminate unfavorable (undockable) molecule configurations in this way without wasting computational resources. The current DD technique may be integrated into existing drug discovery pipelines that use popular docking applications. An automated approach has also been created to help drug-discovery scientists adopt DD, who have little or no familiarity with machine learning and programming.
The Experimental model of Deep Docking (DD)
A chemical library must be preprocessed for DD, just as it must be for traditional docking. Explicit isomeric configurations, proper ionization, tautomers, and protonation states must be provided for each molecule. Then the simplified molecular-input line-entry system (SMILES) of the produced molecules is used to compute circular binary Morgan fingerprints with radius two and a size of 1,024 bits to be utilized as descriptors. These extended-connectivity fingerprints are based on a fixed-length binary bit vector representing the presence or absence of certain substructures and represent a machine-readable description of molecules. The fingerprint bits are employed as features in DNN models designed to figure out which substructures are responsible for high projected binding affinities. ZINC and Enamine’s “make-on-demand” collections are the most widely utilized ultra-large chemical libraries.
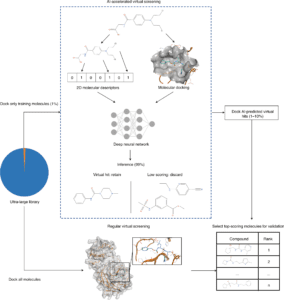
Image Source: Artificial intelligence-enabled virtual screening of ultra-large chemical libraries with deep docking.
Before the docking grids can be generated, the target structure must be prepared. Non-structural water, lipids, and solvent molecules are typically eliminated; structural optimization of the target protein may be required to replace any missing areas, add hydrogens, compute accurate protonation states of residues, and energetically relax the structure. The structure optimization can be done with several licensed and free docking software. Maestro’s Protein Preparation Wizard tool makes such preparation automatic and straightforward. On the other hand, the generation of docking grids entirely depends on the docking application that the user desires to utilize.
At the first DD pass- the validation, test, and initial training sets are randomly selected from the entire docking library. From the second iteration onwards, the training set is continuously augmented with random batches of molecules categorized as virtual hits in the previous iteration’s inference step. The sample size chosen should be large enough to represent the chemical diversity of the library under investigation adequately. At the same time, the maximum sample size is intricately bound to the total amount of docking that the user’s system allows.
Model training and inference are included in each iteration phase of the DD methodology. The protocol employs binary classifiers in feedforward DNN models prepared on 1,024-bit circular Morgan fingerprints to detect virtual hits. In the training, validation, and test sets, binary ‘positive samples’ are virtual hits with scores over a threshold, corresponding to a top preset percentage of the validation set’s docking-ranked molecules. The remaining molecules are designated as ‘negative samples’. The user can specify these top-percentage values for the first and last iterations, but the value will change linearly between those two during the intermediate iterations.
After the generation of binary labels, a grid search technique is used to train a user-specified range of models with various combinations of parameters to improve model test set accuracy. It’s worth noting that as the top-percentage threshold value decreases linearly with the number of iterations, virtual hit calling becomes more stringent; as a result, the definition of ‘positive’ and ‘negative’ labels for all molecules in the training, validation, and test sets changes at each iteration. The inference is always made throughout the entire library, with the beginning percentage value for virtual hit choice set to 1% and the final value set to 0.01%.
“For most docking campaigns, these parameters are sufficient to shrink a database of 1–1.5 billion molecules to a few million compounds that could be conventionally docked with regular computational resources. Alternatively, the preset recall value could be adjusted for more ‘aggressive’ DD-selection of top-scored compounds”, the scientists reasoned.
Any popular docking application can be used in combination with the DD protocol. Using the Fast Exhaustive Docking (FRED), Glide, Autodock-GPU, QuickVina2, and ICM docking suites, we were able to dock billion-size (1B+) chemical libraries against diverse targets in our DD operations. The tools used to prepare proteins, ligands, and docking grids can be easily adapted to similar programs and computer-aided drug discovery (CADD) packages.
One of the most significant issues in modern CADD is the ever-increasing demand for computer resources to screen chemical libraries that are rapidly growing in size due to recent developments in automated synthesis and robotics. Depending on their code scalability over supercomputing clusters, a few docking packages such as OpenEye and GigaDocking have proven successful for screening 1B+ libraries. However, most researchers cannot finance conventional docking of ultra-large libraries due to the high computing costs. As a result, different machine learning emulation docking techniques have been presented to execute such jobs without requiring a lot of computer power.
Although these methods cannot be compared easily (due to the use of various benchmark settings and docking libraries), it is possible to specify that DD is one of the quickest AI-enabled docking systems and the only approach that has been thoroughly examined on 1B+ libraries. Furthermore, the DD protocol is not dependent on a specific docking program; hence it is compatible with emerging large-scale docking methods that aim to increase their high-throughput capabilities.
Story Source: Gentile, F., Yaacoub, J. C., Gleave, J., Fernandez, M., Ton, A. T., Ban, F., … & Cherkasov, A. (2022). Artificial intelligence-enabled virtual screening of ultra-large chemical libraries with deep docking. Nature Protocols, 1-26.
Data Availability: https://doi.org/10.1038/s41596-021-00659-2
Code availability: The DD code is freely available at https://github.com/jamesgleave/DD_protocol
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Swati Yadav is a consulting intern at CBIRT. She is currently pursuing her Master's from Delhi Technological University (DTU), Delhi. She is a technology enthusiast and has a keen interest in the latest research on bioinformatics and biotechnology. She is passionate about exploring new advancements in biosciences and their real-life application.









 driven method that provides very economical yet highly reliable access to ultra-large docking.){kind=link}