
Scientists from the Graduate School of Advanced Science and Engineering at Waseda University, Japan, developed RaptGen, a variational autoencoder for in silico aptamer generation. RaptGen takes advantage of a profile hidden Markov model decoder to address motif sequences efficiently. The model could accelerate nucleic-acid-based therapeutics.
Image Source: Generative aptamer discovery using RaptGen
Nucleic acid aptamers are created by an in vitro molecular evolution technique called systematic evolution of ligands by exponential enrichment (SELEX). Different candidates are restricted by actual sequencing information from an experiment.
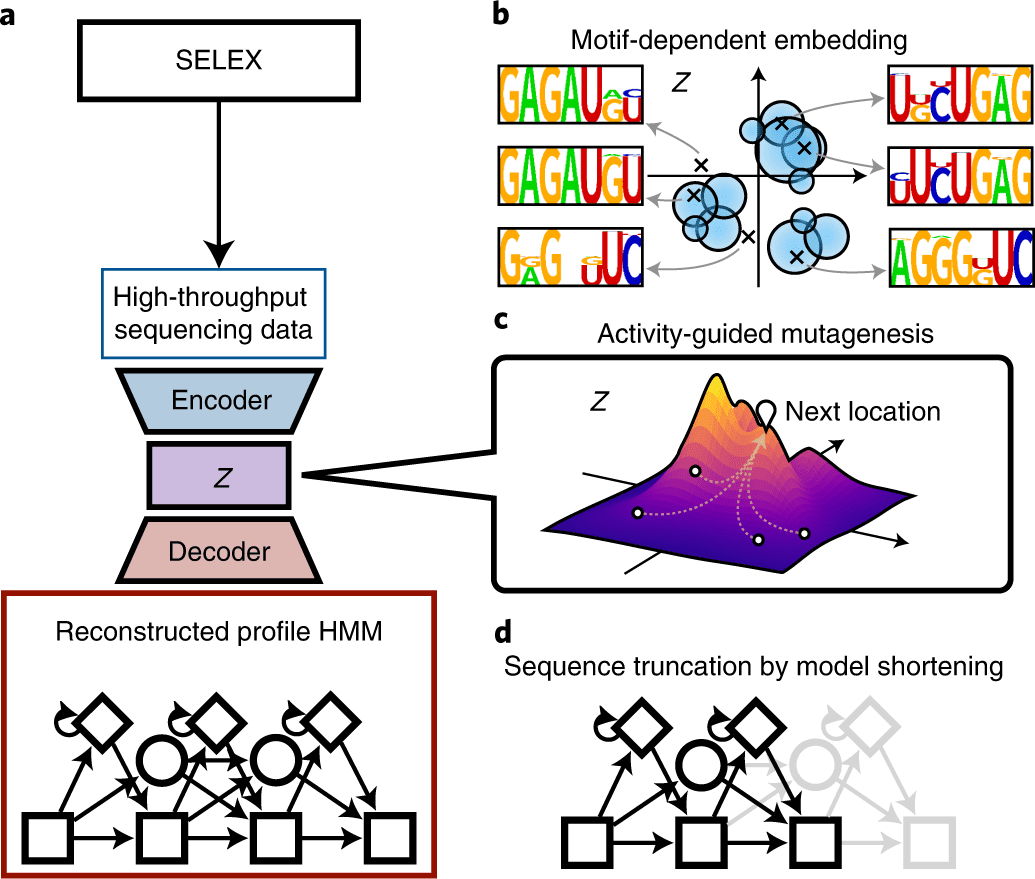
In this study, the scientists explain the developmental cycle of RaptGen, which is a variational autoencoder for in silico aptamer generation. RaptGen takes advantage of a profile hidden Markov model decoder to address motif sequences efficiently.
They showed that RaptGen embedded the simulation sequence information into low-dimensional latent space based on motif data.
The researchers likewise performed sequence embedding by the utilization of two autonomous SELEX datasets. RaptGen effectively produced aptamers from the latent space even though they were excluded from high-throughput sequencing.
RaptGen could likewise produce a truncated aptamer with a short learning model. The scientists showed the way that RaptGen could be applied to activity-guided aptamer generation as per Bayesian optimization. They inferred that a generative strategy by RaptGen and latent representation are valuable for aptamer discovery.
Aptamers: What and Why?
Aptamers are short single-stranded oligonucleotides that bind to explicit targets through their three-dimensional folding structure.
They are known to be analogous to antibodies and have a range of utilizations, including therapeutics, biosensors, and diagnostics. The upsides of aptamers are that they are quickly developed by in vitro generation, are low immunogenic, and have a wide scope of binding targets, which include metal ions, proteins, transcriptional factors, viruses, organic molecules, and bacteria.
Aptamers are produced by the systematic evolution of ligands by exponential enrichment (SELEX). SELEX includes iterations of affinity-based separation and sequence amplification.
This iterative cycle brings about an enriched pool that is analyzed for candidate selection. Latest advances in high-throughput sequencing have empowered scientists to lead high-throughput SELEX (HT-SELEX) to gather an immense number of aptamer candidates.
Current sequencing procedures can assess a limited number of reads: roughly 106 micrograms of a SELEX input library just contains around 1014 duplicates of RNA, whereas an RNA library containing a 30 nt random region theoretically has 1018 (~430) unique sequences.
Consequently, the scientists can assess a tiny portion of the theoretical diversity; hence, computational methodologies that efficiently process high-throughput sequencing information are basic in aptamer development.
Aptamer Identification
A few computational methodologies that recognize aptamers by the utilization of HT-SELEX information have been accounted for. Aptamer identification tools make use of parameters related to the SELEX principle, for example, frequency, enrichment, and secondary structure.
Even though they are valuable for distinguishing sequences from HT-SELEX information, different candidates are restricted by the actual sequence existence in the data.
Simulation-based strategies have been accounted for sequence generation; be that as it may, these techniques require going before motif data and are thus not suitable for the identification of aptamers against an unfamiliar target.
Computational methodologies have likewise been created to predict aptamer motifs. Motif prediction is helpful for candidate discovery as well as for aptamer advancement processes like truncations and chemical modifications.
A few strategies have been created for motif detection by the utilization of secondary structures, enrichment of subsequences during SELEX experiments, and accentuation of different loop regions.
Notwithstanding these methodologies, AptaMut makes use of mutational data from SELEX experiments. As nucleotide substitutions can increase aptamer proclivity, mutational data is useful for candidate discovery.
In any case, even though insertions and deletions are likewise significant elements for altering aptamer activity, in silico techniques that deal with these mutations are inadequately developed; hence, a strategy that creates sequences from experimental information is expected to expand the exploratory space, and including motif data and nucleotide mutations give an expanded opportunity for aptamer discovery.
Neural Networks for Aptamer Generation and Motif Discovery
The scientists zeroed in on neural networks to foster a methodology for aptamer generation and motif finding. As detailed already, neural networks are suitable for the examination of huge datasets and are viable with high-throughput sequencing information.
DeepBind takes on convolutional neural networks (CNN) to recognize DNA motifs from transcriptional factors and finds sequence motifs by the visualization of network parameters. Recurrent neural networks can likewise be utilized for sequence discovery.
Neural network-driven generative models are currently being applied in an expansive scope of research areas. A few instances of neural network-dependent generative models incorporate deep belief networks, variational autoencoders (VAEs), and generative adversarial networks.
For a probabilistic generation of nucleic sequences, utilizing long short-term memory (LSTM) was proposed to imitate sequence distribution. Generative adversarial network-based sequence generation techniques have likewise been proposed.
VAE Based Aptamer Generation Frameworks
Variational autoencoder-based compound designs have been accounted for in small molecule discovery. VAEs gain proficiency with a representation of the information by the reconstruction of the input information from a compressed vector.
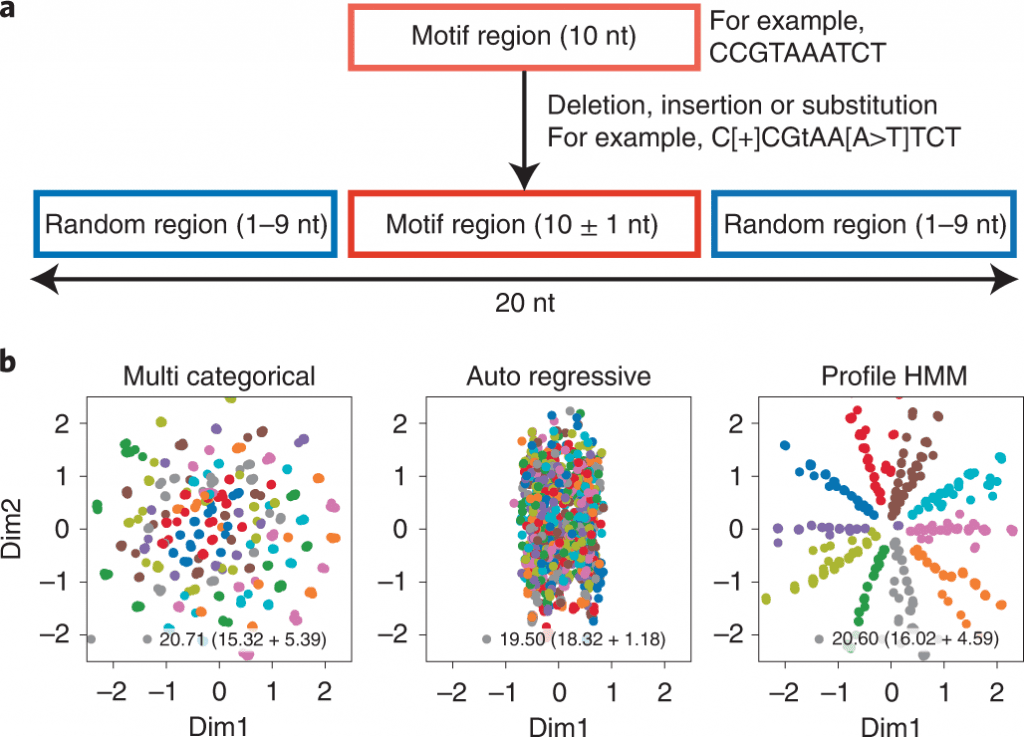
Image Source: Generative aptamer discovery using RaptGen
Not at all like other generative models, VAEs exploit the connection between compressed feature space and inputs in a bidirectional way; they are in this way suitable for the visualization of similarity-oriented classifications and stressing significant sequence features.
Utilizing VAEs to change over HT-SELEX information into low-dimensional space would be helpful for candidate discovery; hence, VAE-based aptamer generation frameworks are worth investigating.
While directing VAE demonstrating for HT-SELEX information, having a profile hidden Markov model (HMM) decoder ought to be valuable for aptamer discovery; it catches motif subsequences — robust with substitutions, deletions, and insertions — and can undoubtedly screen impacts from the subsequences.
RaptGen
In this study, the scientists present RaptGen, a VAE for aptamer generation. RaptGen utilizes a profile HMM decoder to productively make latent space in which sequences structure clusters given motif structure.
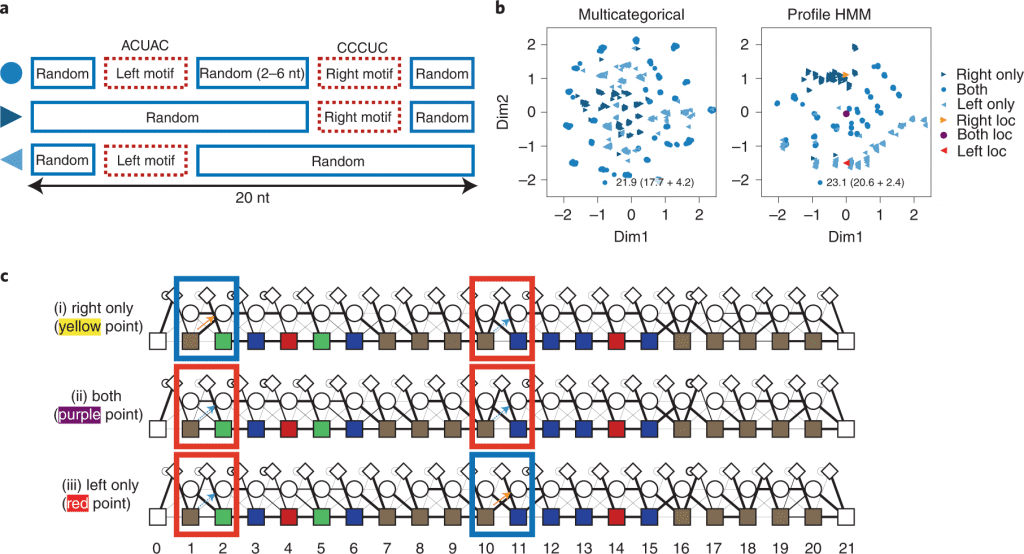
Image Source: Generative aptamer discovery using RaptGen
By the utilization of the latent representation, the scientists created aptamers excluded from the high-throughput sequencing information. Techniques for sequence truncation and activity-guided aptamer generation are additionally proposed.
The Endpoint
In this study, the scientists showed the way that RaptGen could propose candidates as per activity distribution. As per Bayesian optimization, a sequential construction of posterior distribution would permit us to upgrade activity in latent space.
For one more occurrence of the Bayesian optimization application, one could set the securing capacity to different markers other than the limiting activity. We could thus create applicants as indicated by various properties of interest, including inhibitory activity against catalysts or protein-protein interactions. The use of RaptGen for this intention is promising.
RaptGen could progress HT-SELEX information-driven RNA aptamer generation. As an RNA aptamer binds to the target protein by the structural complementarity, not by hybridization, the interaction between the RNA and the protein is barely predicted without binding experiments like SELEX.
When enough of the aptamer-protein pairs and binding information is aggregated, a new aptamer design without wet-lab experiments will be acknowledged from here on out. Moreover, simulation-based strategies, for example, molecular dynamics, will likewise be powerful to work on a computational aptamer design (for instance, aptamer optimization).
Article Source: Iwano, N., Adachi, T., Aoki, K. et al. Generative aptamer discovery using RaptGen. Nat Comput Sci (2022). https://doi.org/10.1038/s43588-022-00249-6
Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.











{kind=link}