
Despite huge leaps being made in the field of gene profiling in recent years, few of the tools developed for single-cell and single-nucleus sequencing take into account spatial variations and positioning, thus leading to the loss of invaluable data. A new method, Slide-tags, aims to remedy this in a manner that is efficient and can be easily integrated with existing computational workflows.
Recent advancements in genomics over the course of the past few years have resulted in the creation of a large number of tools for single-nucleus and single-cell sequencing. However, these toolkits don’t take into account the cell’s cytoarchitectural organization. Some technologies resolve this through barcoding macromolecules whose positions within the cell are known. However, directly transferring design principles to such gene profiling methods is nearly impossible and necessitates the recreation of all molecular assays in a context that takes into account their spatial positions. Further, though the tools available for single-cell sequencing are mature, they require redesigns in order to resolve existing issues with the proper address of cellular noise. A common alternative to such methods is isolating single cells while simultaneously retaining barcoding data – so far, this has only been demonstrated at a limited resolution and sparse tissue samples. Ideally, such a technology would be able to capture cellular profiles efficiently, resolve their positions at high resolutions, and be widely applicable to single-cell methods.
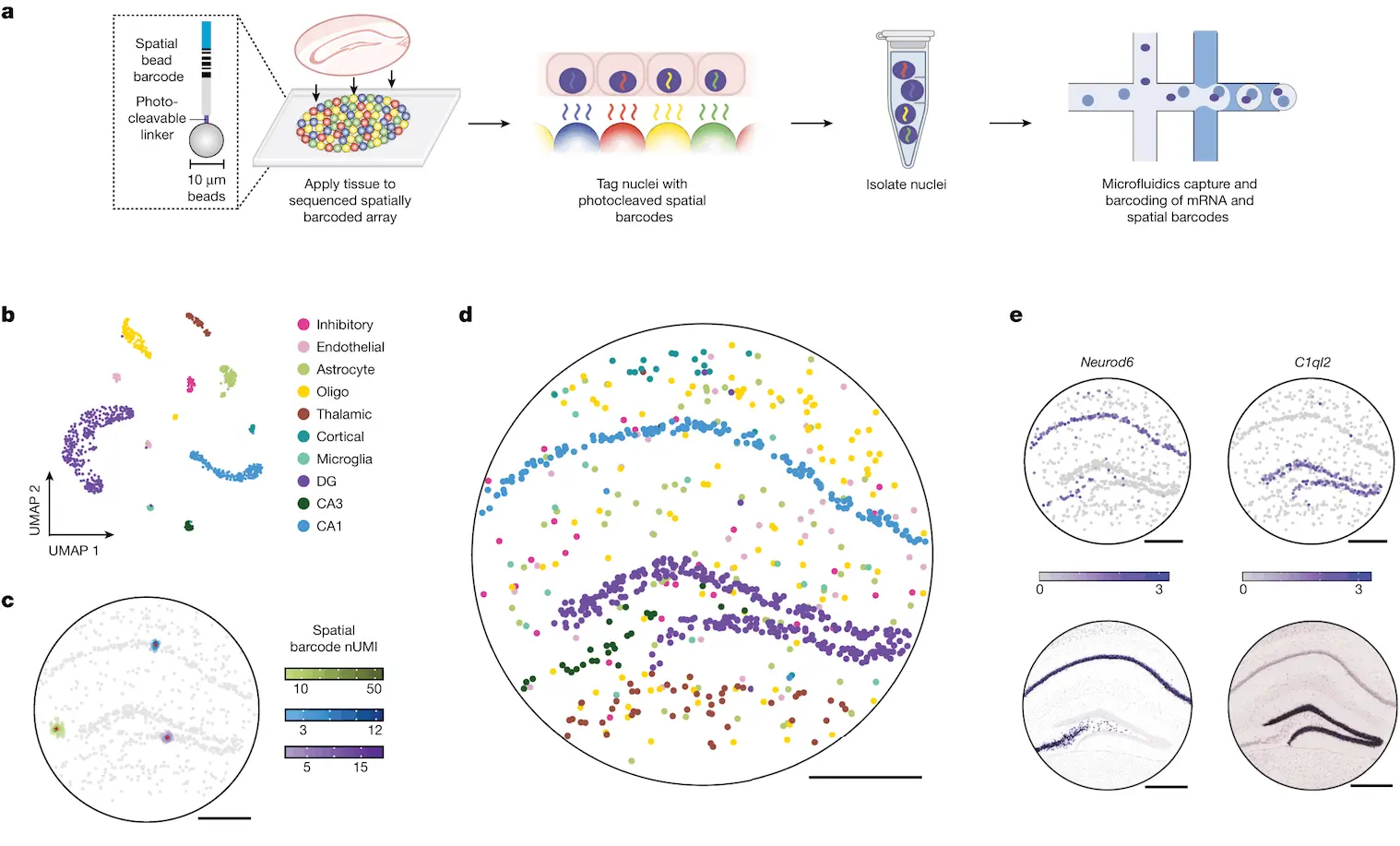
A new method, Slide-tags, aims to resolve these issues: in this method, nuclei from intact tissue sections are barcoded using oligonucleotides. The nuclei are isolated and profiled utilizing existing methods, along with incorporating spatial positioning.
This method can also be easily incorporated into established workflows, like copy number variation inference. The unique characteristics of Slide-tags are leveraged in order to show spatial variations of gene expressions within given cell types, contextualize ligand-receptor interactions spatially, as well as examine the various factors that influence the tumor microenvironment.
DNA-barcoded beads in spatially-indexed arrays were generated utilizing split-pool phosphoramidite synthesis and indexed through sequencing-by-ligation. In the original method, nucleic acids from these tissues were captured and barcoded using such arrays. In contrast, Slide-tags require the photocleavage and diffusion of these barcodes into tissue sections such that they are associated with cell nuclei. Once associated, these barcodes can be utilized as input for established single-nucleus sequencing methods, requiring only minor changes to the protocol.
Slide-tags were performed on a section of a mouse hippocampus, which is known to possess a very stereotyped architecture often used to validate spatial methods. More than 160 nuclei were dissociated and sequenced, and the data was clustered utilizing a standard pipeline, with clusters annotated with established markers. Multiple barcodes were observed per nucleus, thus facilitating greater assignment confidence compared to methods where only a single barcode is associated with each cell. In order to position these transcriptomes spatially, DBSCAN was used to separate the true signal from background noise. The nuclei were then given a spatial coordinate, and in doing so, spatial locations were assigned to more than 800 nucleus profiles.
Examining the positions of clusters resulted in conformance to the expected architecture of the mouse hippocampus. In addition, the expression profiles of individual genes agreed with existing data. To quantify the accuracy of the spatial positioning, Slide-tags results were compared to Nissl images and found to be congruent with each other. In addition, sub-cell types could be accurately localized at different positions within the tissue. Compared to existing methods such as XYZ-Seq and sci-Space, Slide-tags achieved a spatial resolution that was more than 50 times higher and recovered more than four times more nuclei per unit area. Slide-tags also achieved much higher molecular sensitivity than both Slide-seq and DBIT-seq.
When used to profile a section of the human prefrontal cortex, more than 17,000 high-quality nuclei were recovered, with clustering analysis showing glial and neuronal cell types and aligning with known spatial structures. Whole-transcriptome snRNA-seq profiles were used to identify genes that varied spatially according to cell type. The layer distributions were plotted, and well-known laminar markers were recovered. Spatially varying genes were also identified in oligodendrocyte precursors, which weren’t previously known to possess areal specializations. This demonstrates Slide-tags’ ability to reveal transcriptional variations within the tissues present in the human brain.
The method also enables the spatial contextualization of expected interactions between receptors and ligands in human tonsil tissue. The multimodal capacity of the method was also demonstrated through the simultaneous profiling of the transcriptome, epigenome, and TCR repertoire. The copy number alterations could also be inferred using data from the transcriptome and enabled the discovery of spatial differences between cell clones. Transitional tumor states were identified in a cell subclone, and spatial chromatin accessibility data was leveraged so as to identify spatially related motifs that influenced the cell’s transition.
Conclusion
As a novel technology in the field of spatial genomics, Slide-tags offer several different advantages: it can easily be imported into frozen tissue experiments and allow for the addition of spatially resolved data while not necessitating the use of specialized machinery or sacrifices in quality. The method generates data at a single-cell resolution without requiring segmentation and deconvolution and possesses high sensitivity.
Its high-throughput characteristics allow for the profiling of multiple tissue sections simultaneously and is adaptable enough that it can be easily modified to several different existing methodologies. Slide-tags, with further research, could be used to enable the profiling of epigenetic modifications, proteins, and DNA. Though Slide-tags could be improved in some areas, such as nuclei loss and being largely useful for single-nucleus sequencing, its overall utility and adaptability make it an invaluable instrument for research in the field of tissue biology.
Article Source: Reference Paper | Reference Article | Code for processing spatial sequencing libraries is available at GitHub and Zenodo.
Learn More:




Sonal Keni is a consulting scientific writing intern at CBIRT. She is pursuing a BTech in Biotechnology from the Manipal Institute of Technology. Her academic journey has been driven by a profound fascination for the intricate world of biology, and she is particularly drawn to computational biology and oncology. She also enjoys reading and painting in her free time.











{kind=link}