
A research team from Nagoya University in Japan has created a 3-Dimensional computational simulation of the formation of the genome structure in the human cell nucleus. The model is expected to help in understanding cellular regulatory mechanisms and diseases like cancer that modify DNA.
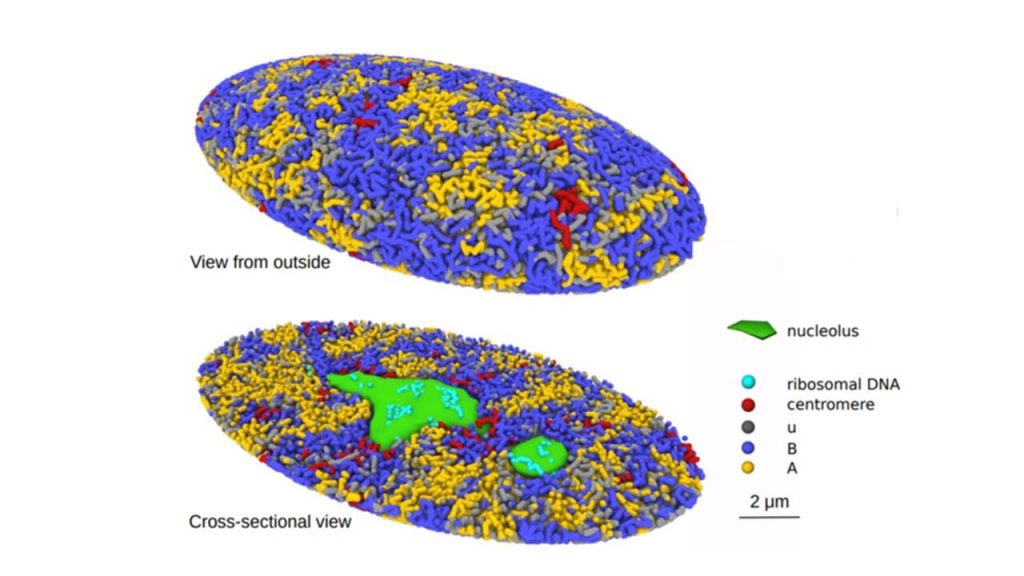
Image Source: https://www.nagoya-u.ac.jp/researchinfo/result-en/2022/06/20220623-01.html
Because it influences DNA reading and replication, the three-dimensional shape of the genome is crucial in controlling how animal and plant cells use their DNA. Our understanding of genome organization has improved as a result of the recent development of tools like high-throughput chromosomal conformation capture (Hi-C and related methods), electron microscopy, and superresolution microscopy. It is essential to create trustworthy computational models that can span these many experimental paradigms to elucidate the mechanisms governing genome organization further.
By studying the entire human cell genome, Shin Fujishiro and Masaki Sasai, Professor Emeritus of Nagoya University’s Department of Complex Systems Science, Graduate School of Informatics, created a 3D model. Using the model as a foundation, they investigated the connection between the structure, dynamics, and functions of the human genome. The research results were published in the Proceedings of the National Academy of Sciences of the United States of America’s online edition.
According to Professor Sasai, the spatial arrangement of DNA and its dynamic activity in cells are crucial for understanding the functioning of the cell. While many experimental techniques, including biochemical and microscopy technologies, have been developed to help researchers understand how DNA is organized in cells, but now a comprehensive picture is required. Thus, the first computational model capable of quantitatively analyzing various data from the complete genome of human cells is presented in this research.
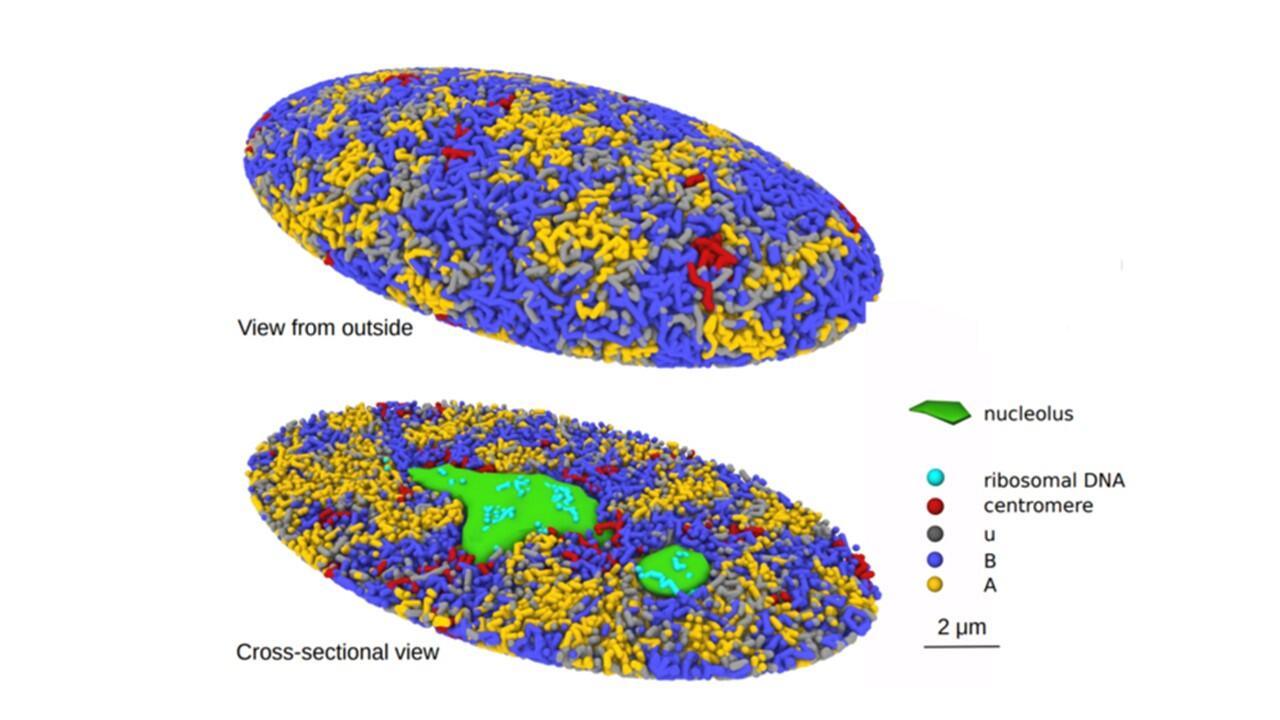
The researchers looked into chromatin to better understand the mechanism. In order to preserve the DNA compact, cells produce chromatin, a mixture of DNA and proteins. In line with their idea, chromatin is misplaced and undergoes an unfolding process during cell division that causes repulsive forces to drive the chromatin chains to split apart. The nuclei’s chromatin is divided into active and inactive compartments by the same force. The researchers discovered that the proposed mechanism clarified biochemical and microscopic findings of prior studies.
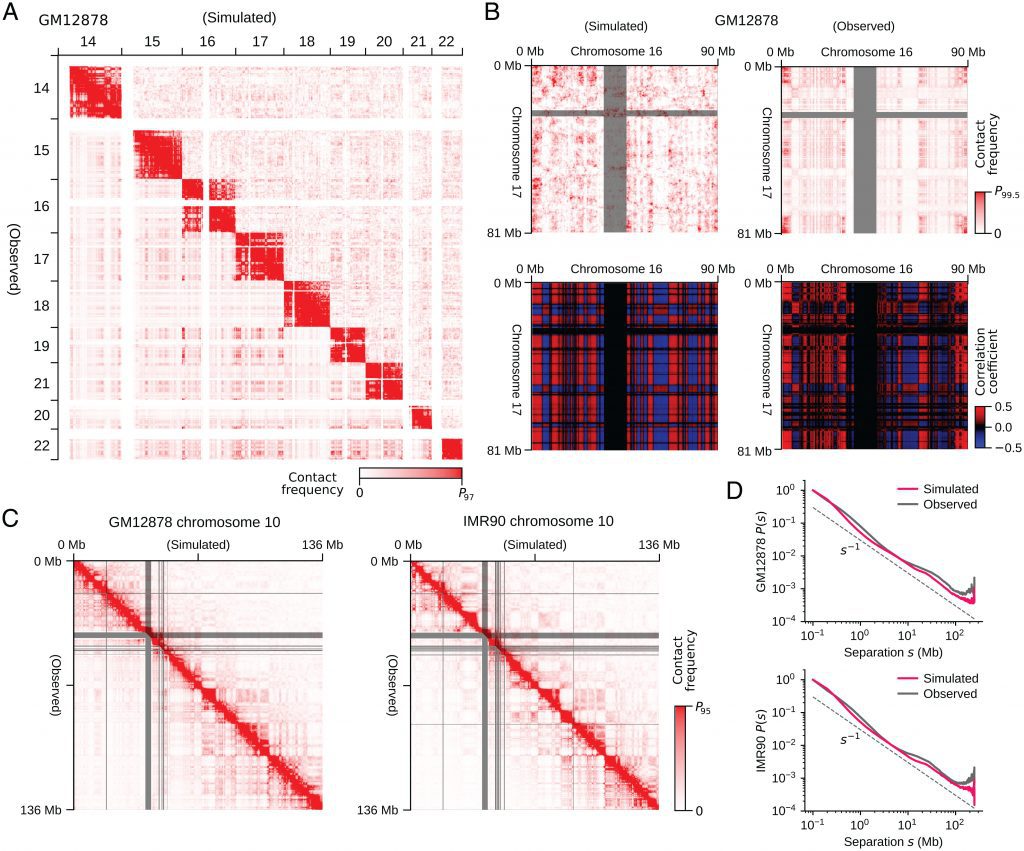
Image Source: https://www.nagoya-u.ac.jp/researchinfo/result-en/2022/06/20220623-01.html
The simulated genome grew upon entering the G1 phase in accordance with the heterogeneous repulsion between chromatin chains, which transported chromatin heterogeneously and caused phase separation of chromatin. This repulsion-driven phase separation accurately predicts the dynamic fluctuations of the nuclei of various human cells and quantitatively replicates the chromatin domains, A/B compartments, lamina-associated domains, and nucleolus-associated domains that have been found in experiments. The researchers suggest that the dynamic genome’s organization is substantially determined by phase separation brought on by diverse repulsive interactions between chromatin chains.
Professor Sasai explains that this model offers a useful resource and a novel viewpoint in cell biology. The model can be used to infer how cell disturbances alter the dynamics and organization of the genome from the computer model created in this study. It can also be used to look into how cancer cells and other illness cells influence the DNA. It enables a more thorough examination of the connection between genomic structure and transcriptional control. The model developed in the research offers a vital tool and a fresh perspective on cell biology.
Story Source: Shin, F., & Masaki, S. (2022). Generation of dynamic three-dimensional genome structure through phase separation of chromatin. Proceedings of the National Academy of Sciences, 119(22), e2109838119. DOI: 10.1073/pnas.2109838119 https://www.nagoya-u.ac.jp/researchinfo/result-en/2022/06/20220623-01.html
Learn More About Bioinformatics:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.







{kind=link}