
USA-based researchers have formulated a supervised deep learning model, ChromaFold, that has achieved state-of-the-art performance in cell-type-specific prediction of 3D contact maps and regulatory interactions only utilizing single-cell ATAC sequencing (scATAC-seq) data, bypassing the prerequisite of multiple input data modalities. Additionally, the lightweight architecture of ChromaFold brings compatibility with standard GPUs; its ability to make cell-type-specific predictions in new cell types, peak-level interactions, and competency to deconvolve bulk chromatin interaction data across constituent cell types facilitate resolving the cell-type-specificity of chromatin interactions.
Unraveling the Genome’s Puzzle: Chromatin Accessibility and Interactions Study
The comprehension of genome architecture and higher-order folding of the DNA is still an unresolved puzzle. The genome confirmation or chromatin arrangement in the nucleus dictates the feasibility and propensity of transcription of a particular gene or genome activity since the physical accessibility of the DNA through dynamism in the chromatin structure determines the opportunity for cellular machinery to extract genetic information. Technically, analysis of the chromatin folds and looping activities enabling proximity between enhancers and genes can reveal the entire landscape and identity of a cell, and such regulatory interactions can also be effectively correlated with the disease-inducing genetic variation that appears in non-coding parts of the genome.
Genome-wide chromosome conformation capture (3C) techniques like Hi-C, HiChIP, and ChIAPET empower scientists to attain such interpretations about physical contacts or adjacency between distant chromatin regions and underlined genetic activity patterns, but due to substantial complexity, high sequencing costs, and difficulty in investigating rare cell types; obstruct the acquisition of high-quality contact maps. Moreover, single-cell chromosome conformation mapping technologies, such as single-cell Hi-C or ChIA-Drop, can capture cell-to-cell variability of three-dimensional genomic features; deliver sparse data sets.
On the contrary, single-cell chromatin accessibility (scATAC-seq) datasets can be readily generated from small amounts of input material using commercially available kits. An assay for Transposase-accessible chromatin using sequencing (ATAC-seq) could unravel the landscape and principles of cellular DNA regulatory variation or accessibility variance, delineating the extent of contact between specific trans-elements, cis-elements such as cell-type specific enhancers and transcription factor binding sites and other nuclear molecules such as histones, chromatin remodelers, and chromatinized DNA. The researchers articulate that, therefore, scATAC-seq information alone can anticipateably assist in predicting chromatin interactions and extrapolate the corresponding connection of regulatory elements to their respective genes via the accessibility profiles.
ChromaFold’s Impact: Advancing 3D Contact Map Prediction in Genomic Research
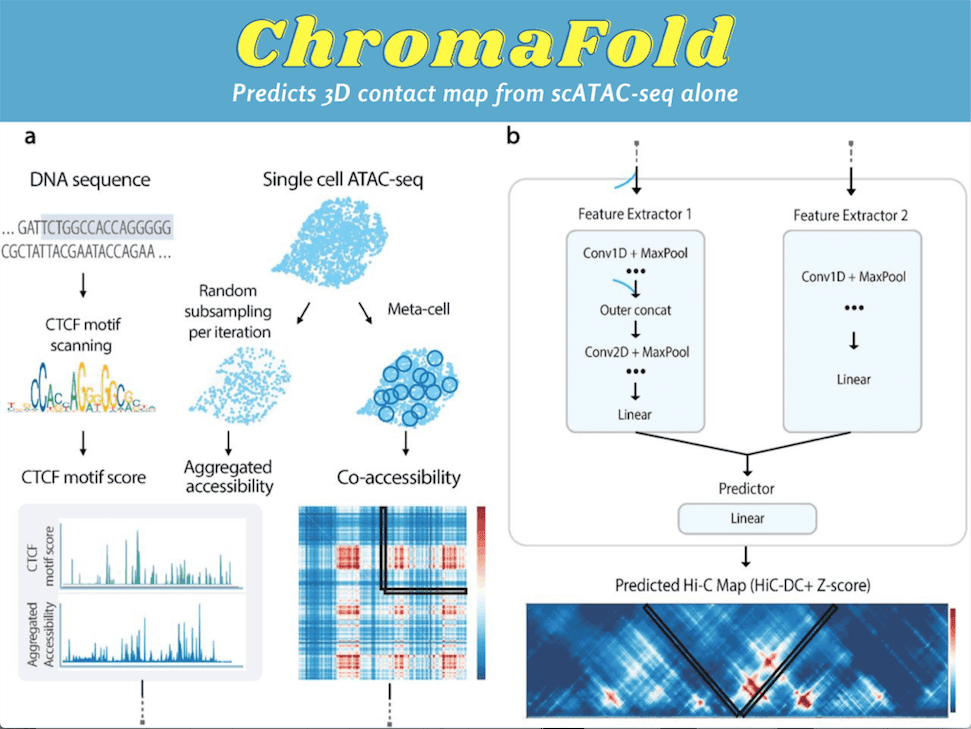
Through ChromaFold, the researchers aspire to remove the requirement of multiple input data modalities for predicting chromatin interactions from genomic sequences. Leveraging supervised Deep Learning, the fundamentals of ChromaFold lie in deriving the linkage between the accessibility landscape of regulatory elements and 3D genome organization. ChromaFold is trained on paired scATAC-seq and Hi-C data from a panel of training cell types.
ChromaFold takes three inputs: pseudobulk chromatin accessibility, co-accessibility profiles across cells, and predicted CTCF motif score/CTCF ChIP-seq. The ChromaFold model architecture possesses two feature extractors and a linear predictor module. The first feature extractor takes the pseudobulk accessibility and the CTCF motif score or ChIP-seq signal as two channels. The pseudobulk chromatin accessibility is obtained by aggregating the accessibility profile across single cells in a population.
This feature extractor comprises fifteen 1D convolutional layers followed by batch normalization and ReLU activation. The second feature extractor takes the co-accessibility data as input. The linear layer consolidates the extracted features and produces a latent representation of the co-accessibility input and the linear predictor to predict the chromatin interactions. A significant improvement was also observed in both the 3D contact map and peak-level interaction prediction when co-accessibility was incorporated as an input, suggesting that the supervised model can extract useful information from the input of the co-accessibility signal.
Furthermore, ChromaFold can not only generalize to new human cell types but also other mammalian genomes, as depicted when ChromaFold was applied for predicted contact maps and peak-level interactions in mouse cells, although it was trained on three human cell types/tissues. Also, it excelled as compared to previous models in terms of robustness and versatility. Apart from these advantageous aspects, several questions are yet to be clarified, such as determining the most relevant epigenomic data achieving good generalization in new cell types, what biological information is captured by introducing a full deep sequence model, etc.
Conclusion
ChromaFold aims to predict chromatin interactions between regulatory elements to their target genes from the scATAC-seq data alone and bring feasibility to comprehending the cell type-specific chromatin interaction landscape where other methods are inconvenient to apply. The witty application of Co-accessibility and CTCF information improves the contact map and peak-level interaction prediction performance. ChromaFold is able to harness cell-type-specific single-cell chromatin accessibility data, make cell-type-specific contact map predictions, and deconvolve chromatin interactions of bulk contact maps in complex tissue or cell populations. It is less susceptible to overfitting and capable of delivering generalizability to new cell types and different mammalian species as well.
Article Source: Reference Paper | ChromaFold’s source code is available on GitHub
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Aditi is a consulting scientific writing intern at CBIRT, specializing in explaining interdisciplinary and intricate topics. As a student pursuing an Integrated PG in Biotechnology, she is driven by a deep passion for experiencing multidisciplinary research fields. Aditi is particularly fond of the dynamism, potential, and integrative facets of her major. Through her articles, she aspires to decipher and articulate current studies and innovations in the Bioinformatics domain, aiming to captivate the minds and hearts of readers with her insightful perspectives.











{kind=link}