
Scientists have developed ColabFold, an open-source software to accelerate the prediction of protein structures as well as complexes by integrating fast homology search of MMseqs2 with AlphaFold2 or RoseTTAFold.
ColabFold offers a sped-up prediction of protein structures and complexes by consolidating the fast homology search of MMseqs2 with AlphaFold2 or RoseTTAFold. The 40−60-overlap quicker search and upgraded model use of the ColabFold empowers the prediction of nearly 1,000 structures each day on a server with one graphics processing unit. Combined with Google Colaboratory, ColabFold turns into a free and open platform for protein folding.
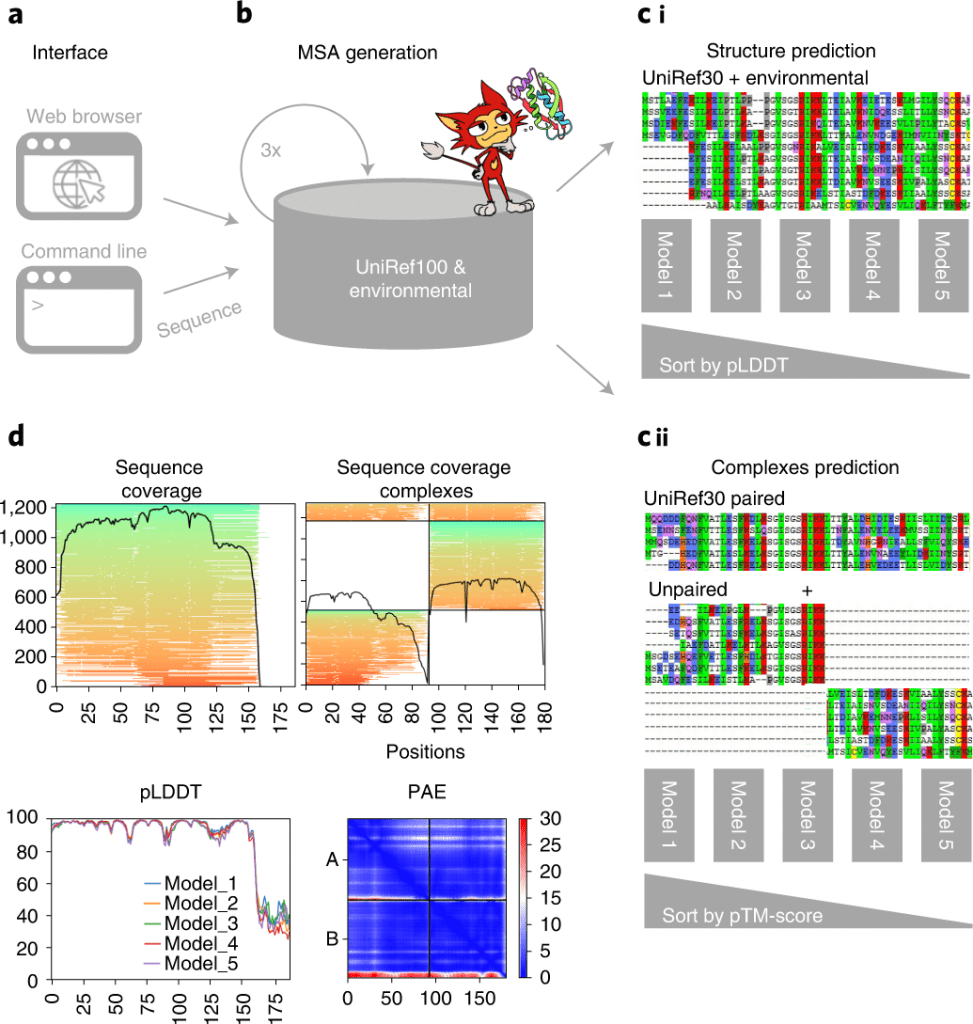
Image Source: ColabFold: making protein folding accessible to all.
An Availability of Highly Accurate Prediction Methods
The three-dimensional structure prediction of a protein from its sequence alone remains an unresolved issue. Be that as it may, by taking advantage of the data in multiple sequence alignments (MSAs) of related proteins as the raw input features for a start to finish preparation, AlphaFold2 had the option to predict the 3D atomic coordinates of folded protein structures at a median global distance test total score (GDT_TS) of 92.4% in the most recent round of the protein folding contest by the worldwide community, CASP14 (Critical Assessment of Protein Structure Prediction, round 14).
The precision of large numbers of the predicted structures was inside the error margin of experimental structure determination techniques. Numerous ideas of AlphaFold2 were freely reproduced and executed in RoseTTAFold.
Notwithstanding predictions for single chains, RoseTTAFold and, later, AlphaFold were also displayed to sum up, protein structures. Scientists have since released AlphaFold-multimer, a refined variant of AlphaFold2 for the prediction of protein complexes. In this way, two exceptionally precise open-source prediction techniques for single chains and one for protein complexes are currently freely accessible.
Overall Run Time Domination of MSA Generation
To use the strength of the strategies, scientists require strong computing capabilities. To begin with, to fabricate diverse MSAs, enormous collections of protein sequences from public reference and environmental data sets are looked through utilizing the most sensitive homology detection strategies, HMMer and HHblits, the two of which use profile hidden Markov models (HMMs).
These environmental data sets contain billions of proteins extricated from metagenomic and transcriptomic experiments, which frequently supplement data sets dominated by isolated genomes. Because of their huge size, searches can require up to hours for a solitary protein while requiring multiple TB of extra storage space alone.
Second, to execute the deep neural networks, graphics processing units (GPUs) with a lot of GPU RAM (random access memory) are required in any event for somewhat average protein sizes of ~1,000 residues. For these, notwithstanding, the MSA generation dominates the general run time.
ColabFold
To empower scientists without the required assets to utilize AlphaFold2, autonomous solutions in light of Google Colaboratory were created. Colaboratory is a proprietary rendition of Jupyter Notebook facilitated by Google.
It is open free of charge to signed-in clients and incorporates access to powerful GPUs. Simultaneously, scientists fostered an AlphaFold2 Jupyter Notebook for Google Colaboratory (alluded to as AlphaFold-Colab), for which the info MSA is built by searching with HMMer against the UniProt Reference Clusters (UniRef90) and an eightfold-decreased environmental data set. This results in less precise predictions while requiring long search times.
In this study, the scientists present ColabFold, a quick and simple to-involve software for the prediction of protein structures and homo-and heteromer complexes, for use as a Jupyter Notebook inside Google Colaboratory, on scientists’ nearby PCs as a notepad or through a command-line interface.
ColabFold accelerates single predictions by supplanting AlphaFold2’s homology search with the 40-60-overlap quicker MMseqs2 (Many-against-Many sequence searching) and speeds up batch predictions by ~90-overlap by keeping away from recompilation and adding an early stop criterion.
The researchers show that ColabFold beats AlphaFold-Colab and matches AlphaFold2 on CASP14 targets, and matches AlphaFold-multimer on the ClusPro dataset in prediction quality.
Parts of the ColabFold Software
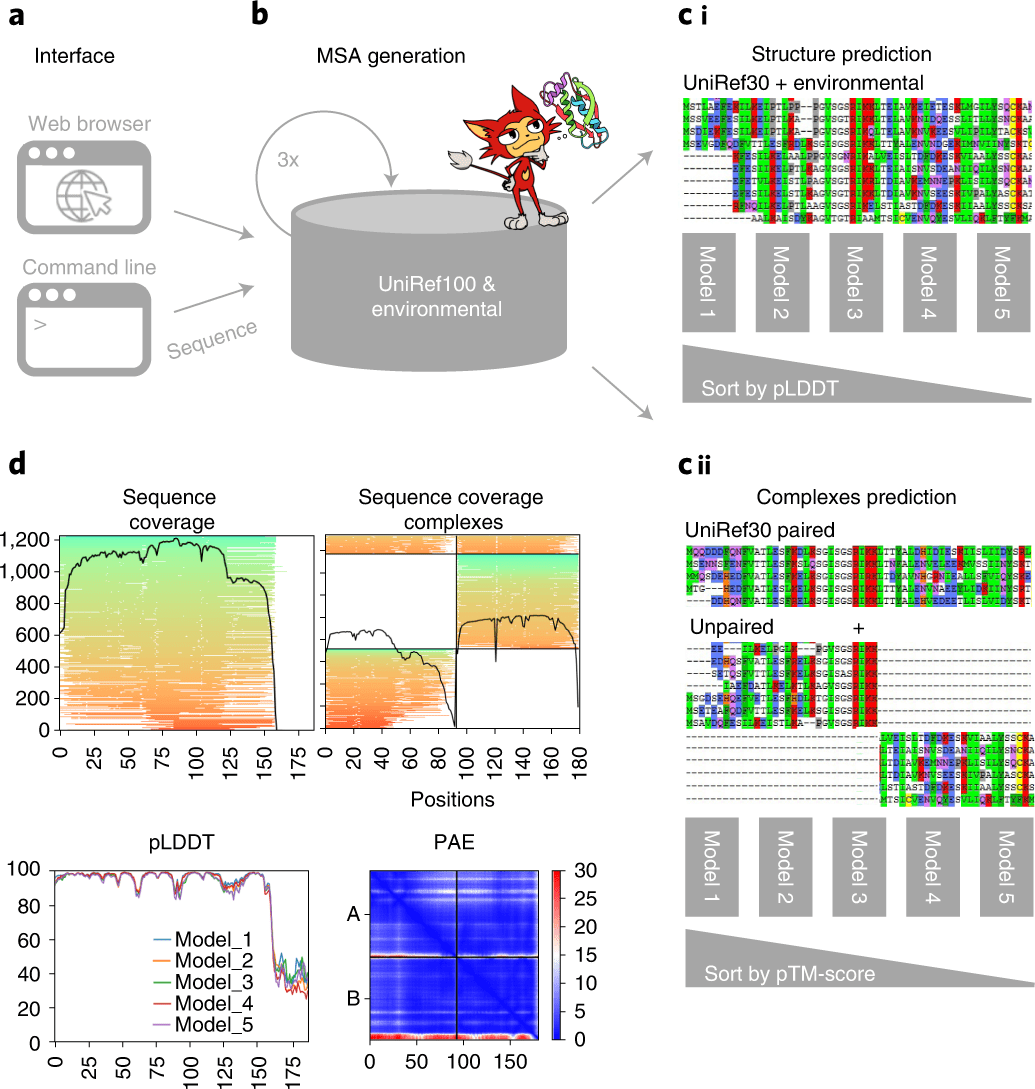
ColabFold comprises three sections. The first one is the MMseqs2-based homology search server to assemble diverse MSAs and track down formats. The server productively adjusts input sequence(s) against the databanks UniRef100, PDB70, and an environmental sequence set.
The subsequent part is a Python library that communicates with the MMseqs2 search server, prepares the input features for structure deduction (single chains or complexes), and visualizes the outcomes.
This library likewise carries out a command-line interface. The last part comprises the Jupyter note pads for fundamental, high level and batch use utilizing the Python library.
Exposing the Internal Parameters of AlphaFold2
ColabFold uncovered numerous interior parameters of AlphaFold2, for example, the recycle count, which controls the number of times the prediction is repeatedly sent through the model.
For troublesome targets as well concerning designed proteins without known homologs, extra recycling iterations can bring about a tremendous high-quality prediction. Rerunning the CASP14 benchmark with a recycle count of 12 brought about an improvement of targets with little MSA data, bringing about an increased average TM-score of 0.898.
For high-throughput structure prediction, the scientists presented a few features in ColabFold. To start with, MSA generation can be executed in batch mode freely from model batch inference.
Second, they aggregate only one of the five AlphaFold2 models and recycle weights. Third, they stay away from recompilation for sequences of similar length.
Fourth, they execute early stop criteria to avoid extra recycles or models if an adequately precise structure was found. Furthermore, last, the scientists fostered the command-line tool colabfold_batch to predict structures on local machines.
All together, they show that the Methanocaldococcus jannaschii proteome of 1,762 proteins more limited than 1,000 amino acids can be predicted in 48 hours with early stopping at a pLDDT (predicted local distance difference test; a for each residue certainty metric) of ≥85 on one Nvidia Titan RTX while forfeiting almost no prediction precision.
The typical pLDDTs of AlphaFold2 and ColabFold Stop ≥ 85 were 89.75 and 88.78 in a subsampled set of 50 proteins.
The Endpoint
ColabFold works past the underlying contributions of Alphafold2 by further developing its sequence search, giving tools for modeling homo-and heteromer complexes, uncovering advanced functionality, extending the environmental databases, and empowering large-scale batch prediction of protein structures at an around 90-crease accelerate over AlphaFold2.
ColabFold is an open-source software accessible on GitHub, and its novel environmental data sets are accessible on Website.
Article Source: Mirdita, M., Schütze, K., Moriwaki, Y. et al. ColabFold: making protein folding accessible to all. Nat Methods (2022). https://doi.org/10.1038/s41592-022-01488-1
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Tanveen Kaur is a consulting intern at CBIRT, currently, she's pursuing post-graduation in Biotechnology from Shoolini University, Himachal Pradesh. Her interests primarily lay in researching the new advancements in the world of biotechnology and bioinformatics, having a dream of being one of the best researchers.











{kind=link}