
A team led by the University of Maryland utilizes machine learning to explain why a "wonder drug" may not work in all patients.
Understanding how mutations cause a medicine to become ineffective is a huge issue. The mechanism by which mutations produce drug resistance is frequently discussed solely in terms of thermodynamics. The case where two proteins have the same drug binding affinities but different residence periods is more confusing.
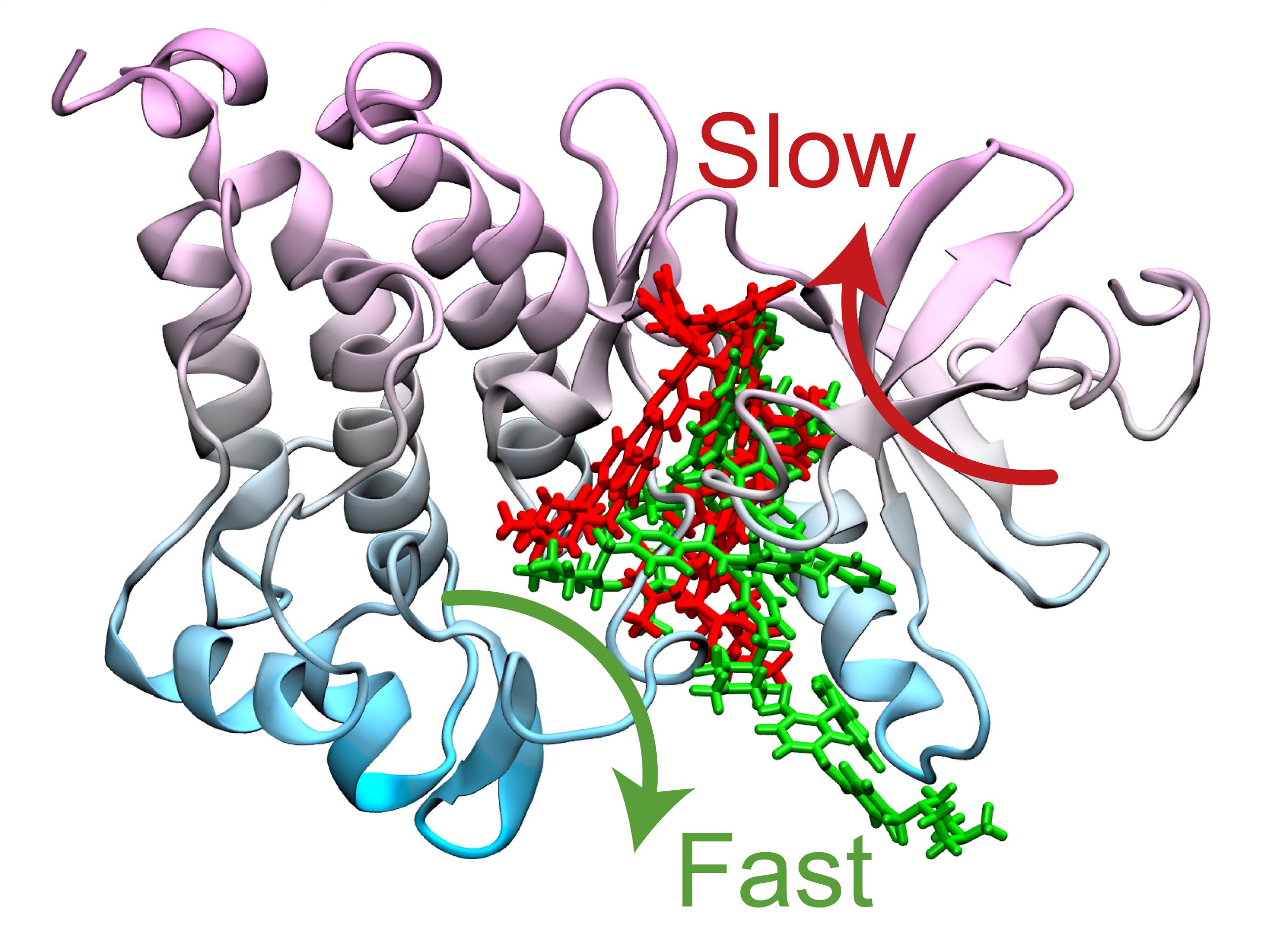
The researchers illustrate how a single point mutation in the protein causes far-reaching alterations in its flexibility, resulting in a new, considerably faster drug dissociation route.
When Imatinib, now known as Gleevec, first hit the market in the early 2000s, it was lauded as a “wonder” cancer medication. Despite its efficacy in treating early-stage chronic myeloid leukemia (CML), a rare malignancy that develops in bone marrow cells, many late-stage patients develop treatment resistance due to changes in key proteins.
A recent study conducted by Pratyush Tiwary, an assistant professor in the Department of Chemistry and Biochemistry and the Institute for Physical Science and Technology at the University of Maryland, used computational chemistry to determine what causes Gleevec resistance at the molecular level.
On April 29, 2022, the study was published online in the journal Angewandte Chemie. It was designated as a ‘Hot Paper’ by the journal’s editors, a designation is given to publications of exceptional interest and significance. The measures created by Tiwary’s lab could have a wide range of uses in the pharmaceutical sector, potentially resulting in more efficacious medications for a number of ailments.
“What this work really does is allow us to figure out which mutations will make any given drug ineffective—and why,” Tiwary said. “Often these could not be predicted just by looking at the structure of a protein. You need these sophisticated simulations that my group does in order to look at this. In physical chemistry, this is known as the thermodynamics versus kinetics debate.”
Tiwary and his co-authors focused on Gleevec, a cancer-fighting drug that works by suppressing a protein called a kinase. The kinase may mutate in some situations as a result of being blocked by the medicine, as a result of environmental conditions, or just by coincidence.
“In some sense, it’s similar to COVID, where mutation happens in the spike protein,” Tiwary explained. “One little part of the protein will change into something else that makes the drug ineffective.”
Drug resistance can be better understood by looking at mutations in the drug-binding pocket, which is where Gleevec binds to a protein. Tiwary and his co-authors, on the other hand, set out to address a more difficult question: why can a mutation called N368S create drug resistance despite the fact that it doesn’t appear to impact the way the drug binds?
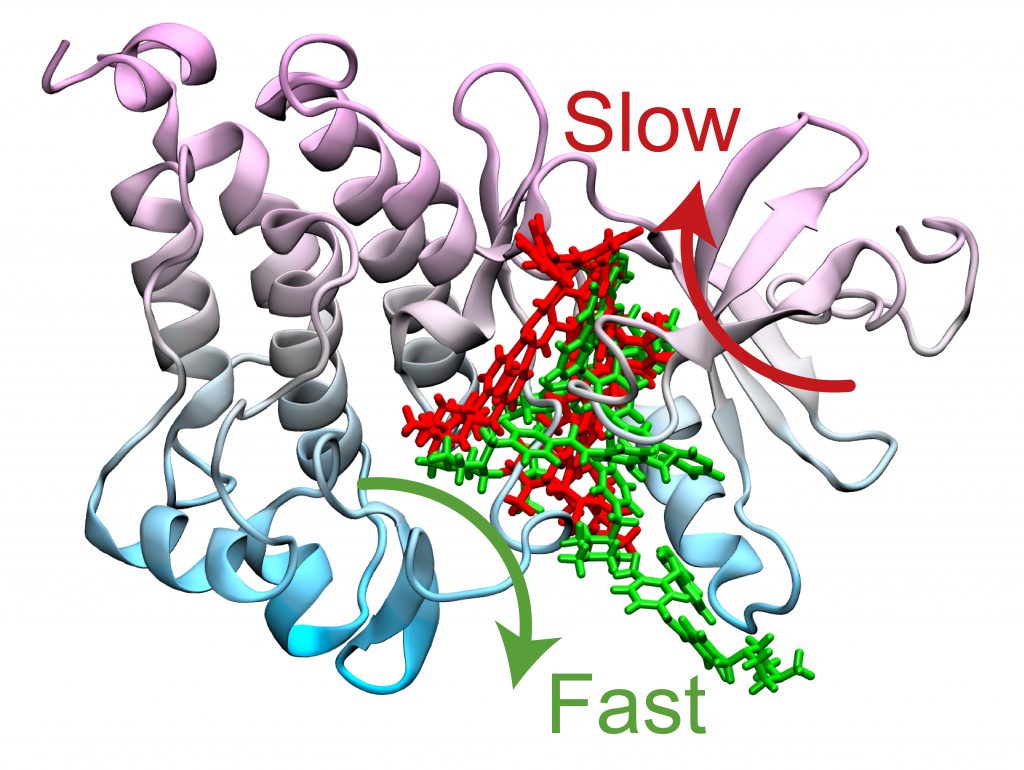
Image Source: https://cmns.umd.edu/news-events/features/4928.
A supercomputer can help with this. The research team employed machine learning and statistical mechanics, a branch of chemistry, to create computer simulation methods capable of investigating incredibly small structures—think atoms and molecules—in extremely high resolution across minutes, hours, and even days. This is unusual because supercomputers can only collect microseconds of data at such high precision in most circumstances.
The model operates by alternating between rounds of machine learning to determine a possible path for Gleevec to depart the protein. This data is utilized to run new simulations, with each round of simulations creating new data that trains the machine learning model to find a better path. The approach is repeated until the machine learning and simulations concur.
The simulations led to the discovery of two optimal paths for Gleevec unbinding from the protein and its N368S mutant. According to Tiwary and his co-authors, because of the N368S mutation, the flexibility of the protein opens up one of these pathways, allowing Gleevec to exit the protein before it starts working.
Tiwary’s strategy might potentially be inverted, with computer simulations used to forecast changes in the drug that would make it more effective and allow it to bind longer, which is one of his lab’s long-term goals. Tiwary’s simulations were employed in another recent study published in ACS Central Science to forecast which mutations would impact the interaction between RNA and small molecules. While computer simulations could never replace traditional drug trials completely, the fact that this new work has been designated as a ‘Hot Paper’ suggests that computational chemistry is gaining traction in the scientific community.
According to Tiwary, for better or worse, there has been a perception in chemistry that simulations aren’t necessarily genuine chemistry. This can be quite ingrained at times. This research demonstrates how simulations can be used to supplement studies in ways that will eventually aid us in tackling challenges of enormous social importance—for cancer and beyond.
This study is an efficient and scalable method for gaining mechanistic insight into biomolecular receptor resistance mutations that are difficult to explain from a structural standpoint.
Story Source: https://cmns.umd.edu/news-events/features/4928
Mrinal Shekhar, Zachary Smith, Markus A. Seeliger and Pratyush Tiwary (2022). Protein Flexibility and Dissociation Pathway Differentiation Can Explain Onset of Resistance Mutations in Kinases. Angewandte Chemie. https://doi.org/10.1002/anie.202200983
Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.











{kind=link}