
In a groundbreaking collaboration spanning IBM Research in Switzerland and the USA, scientists have illuminated the pivotal role of language models (LMs) in molecular discovery. It has been determined that transformer-based architectures are the most popular forms of language models, enabling their widespread use in various domains of science. Due to this, the term ‘scientific language models’ has been coined, indicating language models developed exclusively for scientific research purposes. These models stand as formidable tools, excelling in the domains of de novo drug design, precise property prediction, and intricate reaction chemistry.
The capabilities of language models vary from analyzing small molecules to large macromolecules like proteins and polymers. They have made significant contributions to early-stage drug discovery projects by uncovering promising findings. The researchers outlined the future prospects of designing LM-based chatbots that could possibly access and utilize tools for computational chemistry when prompted to do so. They also listed out open-source resources to increase the accessibility of LMs to individuals who may be interested in working in these domains.
Underlying issues with conventional molecular discovery methods
It is estimated that it can cost upwards of 3 billion dollars for each new drug or pharmaceutical product that will be released in the market over the course of the next ten years. This can be attributed to the expenses involved in performing drug screening in vitro, the increased occurrence of unexpected discoveries, and the vast expanse of chemical data that needs to be screened for the identification and prediction of drugs.
Another issue is that applying traditional scientific methods to molecular discovery is challenging. The ‘Design-Make-Test-Analyse’ (DMTA) is a workflow pattern commonly used by chemists and scientists involved in chemical research, but in many instances, it has hindered the revision and refinement of hypotheses that may include biases of the scientists that proposed them, in turn reducing the flexibility of incorporating more relevant factors into the hypothesis.
Deep generative models can solve this issue. When added to the DMTA workflow, they can refine the hypotheses entered as input for the model while filtering out biassed takes, thus selecting the ones with the most potential to move forward with further steps in the experimentation process.
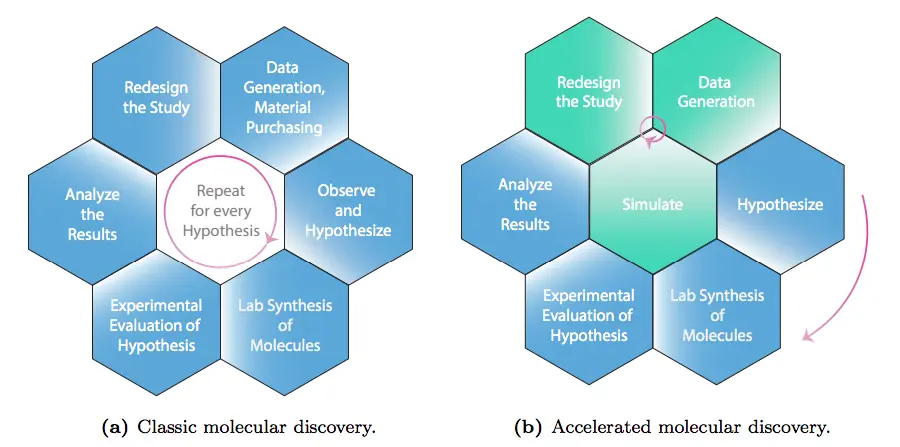
Image Source: https://doi.org/10.48550/arXiv.2309.16235
A brief introduction to language models
Language models have been defined by their abilities to understand context and develop reasoning in relation to the ‘tokens’ that have been fed to them. ‘Tokens’ can be described as a series of texts with a specific function or purpose. Learning the probability distributions throughout these sequences of text has equipped these models with the ability to successfully perform tasks requiring contextualization and can generate texts from their end as output.
The need for language models in molecular discovery
Language models possess the capacity to learn highly structured representations that are specific to desirable functional properties. When they are applied to a series of texts (for example, SMILES) that represent chemical entities, they smoothly make predictions that properly account for all the relevant properties of the protein or molecular entity in question.
These models can also easily form connections between natural and scientific languages, overcoming many barriers. This particular feature gives researchers further scope to develop chatbots that are similar to ChatGPT in the future. Developing such interactive models will facilitate chemists and scientists to perform complex tasks more efficiently, as they can utilize LMs to curate the objectives of any upcoming project with greater clarity while improving the quality of the final results obtained. The authors of the review also highlighted the benefits that come with combining language models that can generate and predict molecular data, respectively.
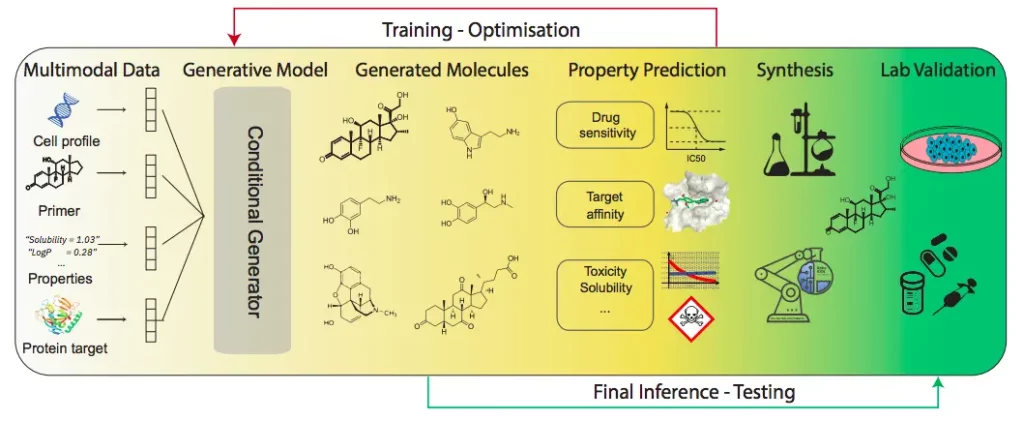
Image Source: https://doi.org/10.48550/arXiv.2309.16235
Various molecular representations and forms exist in literature. It is crucial to identify the most ideal representation that can be used as input for the language model. The components of language models utilized for molecular discovery are given by the following:
1. Simplified Molecular Input Line Entry System (SMILES): It is a representation consisting of strings that incorporate specific characters such as molecular rings, the aromaticity of molecules, the nature of bonds, and other aspects of their stereochemistry. A single molecule can be represented by multiple types of SMILES strings.
2. Tokenization: Here, strings are split into small units to form vectors that can be processed by the language model. These units can range from taking the form of singular characters to full-fledged words. SMILES strings are broken down to an atomic level to create these vectors.
3. Vocabularies: They act as dictionaries that form links between tokens and vectors.
4. Self Referencing Embedded Strings (SELFIES): It is an alternative to SMILES strings for solving the issue of invalid molecules generated occasionally by the latter. They use derivation rules for generation to account for valence-bond validity. They take the length of the branches and the size of the rings into consideration, preventing the possibility of open branches and rings being included in the input of the language model. This ensures 100% validity.
5. International Chemical Identifier (InChI): It contains structural information about molecules arranged in a hierarchical pattern within the string. It has been developed by the IUPAC (International Union of Pure and Applied Chemistry).
A few open-source software provided by the authors
- Natural Language Models
It is a library of transformers created by HuggingFace. A few examples of such models are as follows:
- ChemBerta: Used for predicting molecular properties
- MolT5: Used for molecular captioning and for generating molecules based on the text input
- Multimodal Text and Chemistry T5: Multitaskers that act based on the prompts given to them.
- GT4SD – Generative Modelling Toolkits
The Generative Toolkit for Scientific Discovery is a collection of Python libraries meant for application in molecular discovery. It provides access to graph generative models like TorchDrug and diffusion models.
It is a collection of language models for analyzing the reactions and synthesis of molecular compounds. It constitutes the API of the IBM RXN for Chemistry platform.
Conclusion
Until recently, we never thought it would be possible to ask AI model queries like we usually would for search engines, expecting a well-curated response from them. While AI models currently lack the ability to reason on their own as humans do, they are capable of learning from us. This has led to the establishment of large language models (LLMs) like GitHub Copilot and ChatGPT.
The rise of language models has opened up a collaborative platform to facilitate interactions between experienced scientists and individuals who do not have a scientific background or experience. It enables both classes of people to utilize these models for research without having to undergo specialized training, owing to the ease with which chatbots can be used.
Article source: Reference Paper
Important Note: bioRiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Swasti is a scientific writing intern at CBIRT with a passion for research and development. She is pursuing BTech in Biotechnology from Vellore Institute of Technology, Vellore. Her interests deeply lie in exploring the rapidly growing and integrated sectors of bioinformatics, cancer informatics, and computational biology, with a special emphasis on cancer biology and immunological studies. She aims to introduce and invest the readers of her articles to the exciting developments bioinformatics has to offer in biological research today.











{kind=link}