
Researchers from MIT University, Cambridge, have introduced a novel generative model, ‘SQUID’, to facilitate the shape-conditioned generation of chemically diverse molecules for drug design in 3D conformation. The team has successfully introduced equivariant deep working models to challenge current 2D generative models.
Introduction
Computer-aided drug design, CADD, has been reformed by integrating generative models. CADD has allowed these models to accustom efficient exploration of chemical space exploration, molecular optimization, and constructing virtual chemical libraries. In recent years, several generative models have surfaced, such as convolutional neural networks (CNN) for 3D bioactive molecular conformation, autoregressive models, and diffusion models.
Adams et al. have rendered various loopholes, wherein models have often overlooked these conditions while training models such as the structure of protein targets, presumed knowledge of binding sites of potential drug targets, ignoring the possibilities of 3D protein pocket flexibility, which often are inefficiently addressed in CADD-based settings. The virtual screening methods deployed on the ligand-based drug designing identify the active chemical molecules on previously identified 3D pharmacophores or molecules with similar structures. These methods do not pin knowledge of protein structure or assembly into consideration.
In order to elucidate the potential of novel chemical molecules for exploration in drug designing, researchers from the Massachusetts Institute of Technology, Cambridge, have introduced a new generative model, SQUID (Shape-Conditioned Generator for Drug-Like Molecules). The novel shape-conditioned 3D generating model visibly halts the limitations associated with virtual screening methods and presents different challenges which were not previously addressed in the classical 2D generative models.
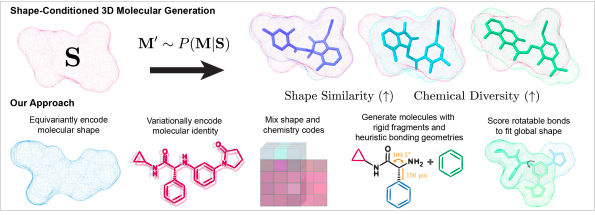
Image Source: https://arxiv.org/pdf/2210.04893.pdf
Some of the challenges addressed by SQUID are:
1) The classical conditioned generative models utilized single scalar property to optimize the data distribution, whereas the SQUID model has coped with this by involving an equivariant point cloud network to give 3D molecules with target shape using GNN (graph neural networks) and a pairwise comparison between the two random conformations of the arbitrary molecules.
2) Unlike 2D similarity, that computed shape similarity between the potential molecules without addressing the sensitivity of molecule conformation. In order to reduce its impact, the researchers have encoded a model with SE(3) invariant representation to generate an aligned model to construct chemical molecules befitting the target shape.
3) Considering the dependability of 2D graph topology and 3D structure, slight changes in the graph can change the access points for the target hence, SQUID reliably generates chemically diverse molecules using a sequential fragment-based 3D generation procedure to anchor the local bond lengths and prioritize the rotatable bonds to simplify the 3D molecule conformation.
4) The inflexibility of large molecular atoms to generate the flexible drug molecules to sustain the steric hindrance and maintain the validity as a drug; thus, the Shape-Conditioned Equivariant Generator overcomes it by designing a rotatable bond scoring network to understand the 3D conformation and align molecules to the understanding of target and enables the decoder to generate the suitable fit.
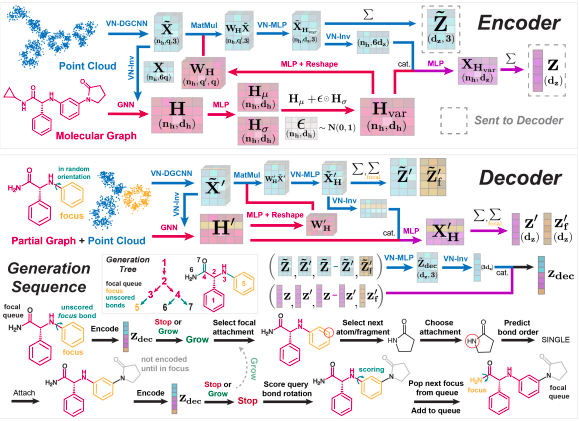
Image Source: https://arxiv.org/pdf/2210.04893.pdf
The methodology of the entire SQUID model was based on the encoder-decoder architecture that generates molecules atom-by-atom, fragmenting them and repetitively growing the molecular graph and generating the 3D coordinates by scoring a network of rotatable bonds. The encoder is constructed by dividing the approach into five fragments that are featurization, fragment encoder, shape encoder, variational graph encoder, and mixing shape and variational features with each invested in fixing bond angles, specifying fragments, and providing to the decoder on an equivariant latent representation. The decoder further divides the sampling of new molecules by dividing it into three skeptical procedures that are partial molecule encoder, graph generation, scoring rotatable bonds, and training the model.
During the experimentation on the efficacy of the model, the researchers trained SQUID with 27 drug-like molecules from MOSES ((Polykovskiy et al., 2020) and obtained a total of 100 fragments and 24 atoms from the dataset. A 3D conformer was constructed for selected fragments using RDKit and conditioned these chemically diverse molecules by imitating the technique of ‘scaffold hopping’ to generate 3D shapes with a high similarity index. The similar drug-like molecules are later optimized by SQUID, impersonating the shape-constrained MO setting from GaucaMol to the benchmark model using open-source Python code for De Novo Molecular Design (Brown et al.). In 40/38 tasks, SQUID improves the object scoring with successful cases of virtual screening to about 1M samples, which were earlier limited to 31K samples, describing its ability to optimize conditions as required to screen and befit the target shape while screening.
Final Thoughts
Kier Adam and team have worked to dissipate the earlier models by introducing a shape-conditioned generator for 3D molecules and reliably generating valid drug molecules. The model SQUID ensured local geometric and chemical validity, which was lacking in other generators, was addressed and optimized by demonstrating its effectiveness in overall virtual screening of constructed libraries. The model has the potential to accelerate the drug-designing process and provide novel hits for pharmaceuticals. While ethical usage and reasoning will be helpful to not exploit the model beyond repair.
Article Source: Reference Paper | Code Availability: SQUID
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Mahi Sharma is a consulting Content Writing Intern at the Centre of Bioinformatics Research and Technology (CBIRT). She is a postgraduate with a Master's in Immunology degree from Amity University, Noida. She has interned at CSIR-Institute of Microbial Technology, working on human and environmental microbiomes. She actively promotes research on microcosmos and science communication through her personal blog.





{kind=link}