
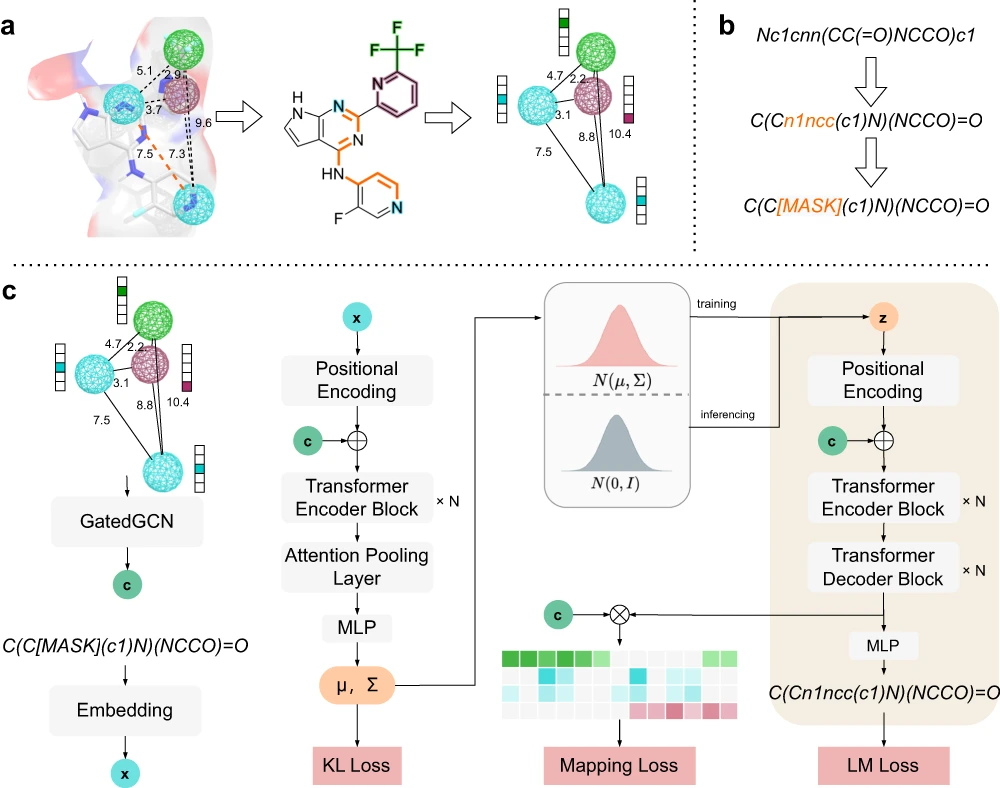
One of the biggest challenges to drug discovery is designing molecules that express desired biological activity, also referred to as bioactivity. This is of great importance when intending to use the drug for treating diseases that have not been thoroughly researched. To tackle this, researchers from Central South University, China, developed PGMG, which stands for ‘Pharmacophore-Guided deep-learning approach for bioactive Molecular Generation.’ It is a flexible model. Graph neural networks are utilized for encoding to distribute chemical features in the deep-learning space. For decoding, transformers are used; this leads to the generation of the desired molecules. The diversity of these molecules is improved by the introduction of latent variables that solve the issue of carrying out large-scale mapping between pharmacophores and generated molecules. This model has vast potential for fine-tuning the drug discovery process.
There are just too many compounds to screen!
Within the expansive chemical space, those compounds that exhibit the properties of pharmaceutically active drugs are determined by analyzing molecular properties based on Lipinski’s ‘Rule of Five,’ according to which a molecule with a molecular mass less than 500 Da, no more than five hydrogen bond donors, no more than ten hydrogen bond acceptors, and an octanol-water partition coefficient log P not greater than five is taken into consideration. The number of such bioactive compounds is estimated to be as large as 106 as of today. This creates a herculean task when attempting to search for desirable drugs for a disease.
Deep-learning methods provide an edge over traditional methods
Until recently, those compounds that displayed even minimal effect on target cells were produced by medicinal chemists in a laboratory setting or were screened from a set of compounds using high-throughput screening. Both of these methodologies have been demanding, time-consuming, and financially exhaustive.
With recent advancements in the fields of artificial intelligence (AI) and machine learning (ML), deep-learning (DL) ‘models have the potential to perform the same tasks without limitations, with much more ease and convenience for researchers. Some of the most popular deep learning models for drug design have used architectures based on autoencoders, autoregressive models, and reinforcement learning.
A lot of the models mentioned above seek to generate molecules based on physicochemical properties that have been specified beforehand, such as:
- Wildman-Crippen partition coefficient (logP): It is an indicator of the ability of the molecule to mix with an oil phase, also known as its lipophilicity
- Molecular weight (MW)
- Synthetic Accessibility (SA)
- Quantitative Estimate of Drug likeness (QED)
However, these models have failed to design molecules that account for properties such as structure-activity relationships and bioactivity, which usually require a substantial number of calculations and vast knowledge of pre-existing molecules; this may not necessarily be available when dealing with rare diseases. The lack of availability of sufficient data is being looked into by using DL models for molecule generation, incorporating previous knowledge of various biochemical properties.
A look into PGMG
Before getting an insight into the workings of the model, it is important to understand exactly what pharmacophores are. According to the IUPAC (International Union of Pure and Applied Chemistry), pharmacophores are ‘a collection of steric and electronic characteristics necessary for ensuring optimal supramolecular interactions with a specific biological target and to block its biological response’. Complicated, right? In simple words, it is a set of chemical features that determine the binding of a drug to a defined target.
The dataset of PGMG uses hypotheses based on biologically meaningful pharmacophores to form an interconnection between several data points on the activity of molecules. The hypotheses can be formulated in two ways:
- Superimposition (placing one thing over the other) of a few active compounds
- Using the structure of the target as a basis for designing drugs
REALTION is an example of a previous model that used pharmacophores, although it only used it as additional support for the purpose of generating molecules that are primarily based on the characteristics of complexes and active molecules. It did not provide enough room for flexibility, limiting its applications.
PGMG is a much more flexible model. Here, pharmacophores are represented by complete graphs; these are the only inputs used for the model; no activity data is required. The features of the pharmacophore correspond to nodes within the graph, where the distance between each node pair gives information on its spatial features, and the graph contains edge attributes as well. Molecules are generated based on the pharmacophore entered as input.
The model can choose from a variety of datasets related to the activity of molecules to form a uniform representation for the regulation of the molecular design process. The transformer component acts as the backbone for learning the implied rules of the Simplified Molecular Input Line-Entry System (SMILES) strings, a group of line notations used for describing the structure of chemical species used in the mapping. Latent variables used in the model help in the characterization of pharmacophores in a unique manner, and they take care of the many-to-many mapping relationships between molecules and pharmacophores.
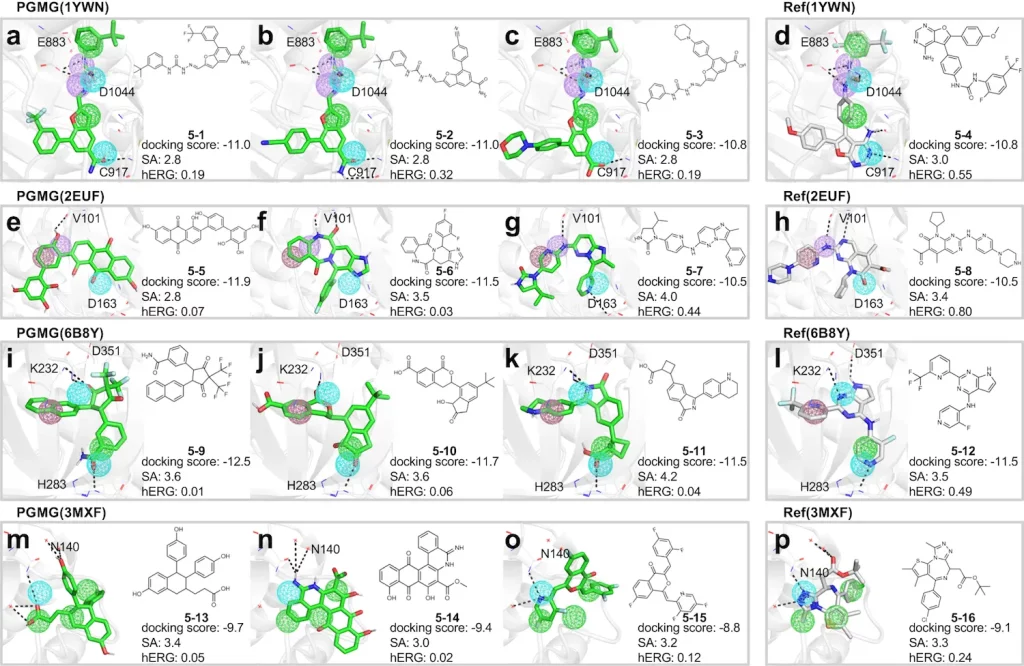
Molecules generated using PGMG show high validity and exclusivity, all while fulfilling pharmacokinetic requirements that are adequately in line with the pharmacophore hypotheses provided beforehand. They have also been shown to have docking scores that are equivalent to, if not better than, those of molecules extracted from the ChEMBL (a manually curated database containing bioactive compounds). PGMG can also be utilized for designing molecules with dual or multiple targets.
Scope for future improvements and conclusion
PGMG can be used for preparing chemical libraries for virtual screening purposes; this will improve efficiency by providing candidate drugs that correspond to a target that has already been specified. It can also examine multiple drugs at the same time, which is a desirable feature that can be applicable to research carried out in the fields of drug resistance and alternative medicine.
De novo drug design refers to methods that create new chemical entities based on information related to a target and its corresponding active binders; it can get complicated as it is a specific methodology. PGMG can be advantageous in this domain as the pharmacophore hypotheses are created by users. It can be altered and adjusted by them to improve accuracy. This can be done through the application of Quantitative Structure-Activity Relationships (QSAR) methods. It will deal with a limitation of PGMG, that is, its inability to put restrictions or constraints over the properties of molecules generated using it, also known as exclusion volume. Future improvements aim to include exclusion volume and to execute greater control over generated molecules. The incorporation of QSAR can improve the interpretability of the model in the long run.
Article source: Reference Paper | PGMG source code is available on GitHub
Learn More:




Swasti is a scientific writing intern at CBIRT with a passion for research and development. She is pursuing BTech in Biotechnology from Vellore Institute of Technology, Vellore. Her interests deeply lie in exploring the rapidly growing and integrated sectors of bioinformatics, cancer informatics, and computational biology, with a special emphasis on cancer biology and immunological studies. She aims to introduce and invest the readers of her articles to the exciting developments bioinformatics has to offer in biological research today.











: A New AI Approach to Drug Discovery){kind=link}
[…] Pharmacophore-Guided Deep Learning: A New Paradigm for Generating Bioactive Molecules in Drug Discov… […]