

Tufts University researchers have developed MELISSA, a groundbreaking approach for predicting protein functions through protein-protein association networks. Existing methods such as Mashup and deepNF employ network diffusion analysis and structure embedding of proteins, respectively, but they neglect to utilize known protein functions directly. Contrarily, MELISSA, a novel approach, incorporates functional labels during the embedding stage. Guiding semi-supervised dimension reduction with these labels enables MELISSA to produce embeddings that encapsulate network topology and functional information. When it comes to predicting Gene Ontology labels and constructing functionally enriched protein neighborhoods in multiplex association networks for yeast and humans, this method surpasses Mashup and deepNF in performance.
An Overview of Protein Function Prediction Methods
In 2016, Cho et al. introduced the Mashup algorithm for biological function prediction using information from multiple biological networks. Mashup involves three steps: using the Random Walk with Restart (RWR) algorithm to compute diffusion state vectors for each node of each network, creating a low-dimensional embedding to capture network-specific information, and applying classifiers for function prediction.
However, biological knowledge (GO functional labels) is only incorporated in the final step of the process, and the embedding is derived solely from network topology. Similar is the case with deepNF, another method that uses a multimodal deep autoencoder for the embeddings.
Researcher Lenore Cowen and his team have introduced MELISSA (MultiNetwork Embedding with Label Integrated Semi-Supervised Augmentation), a novel algorithm aimed at enhancing protein function prediction by integrating known biological knowledge with network-based information. Building on the Mashup algorithm’s foundation, MELISSA tackles the challenge of accurate functional label prediction by introducing a semi-supervised approach early in the embedding process. This involves incorporating “must-link” (ML) and “cannot-link” (CL) constraints, informed by Gene Ontology (GO) functional labels, to guide the embedding procedure in capturing both network topology and functional information.
Notably, the algorithm addresses the complex nature of gene function annotations, characterized by noise and incompleteness, by augmenting networks with artificial nodes representing cluster centers for coarse functional groupings. These nodes are linked to original genes through ML constraints, fostering accurate clustering, while CL constraints encourage cluster separation. The method employs a signed graph Laplacian to handle networks with both positive and negative weights, and the rest of the Mashup pipeline, encompassing dimension reduction and function prediction using the low-dimensional embedding, remains intact.
Pairing MELISSA with the 𝑘-nearest neighbors (𝑘NN) classifier demonstrates its superiority in functional label prediction over Mashup and deepNF embeddings. Ultimately, MELISSA presents a scalable framework for effectively integrating and analyzing diverse biological networks, offering a promising avenue for accurate protein function prediction that leverages both network structure and biological knowledge.
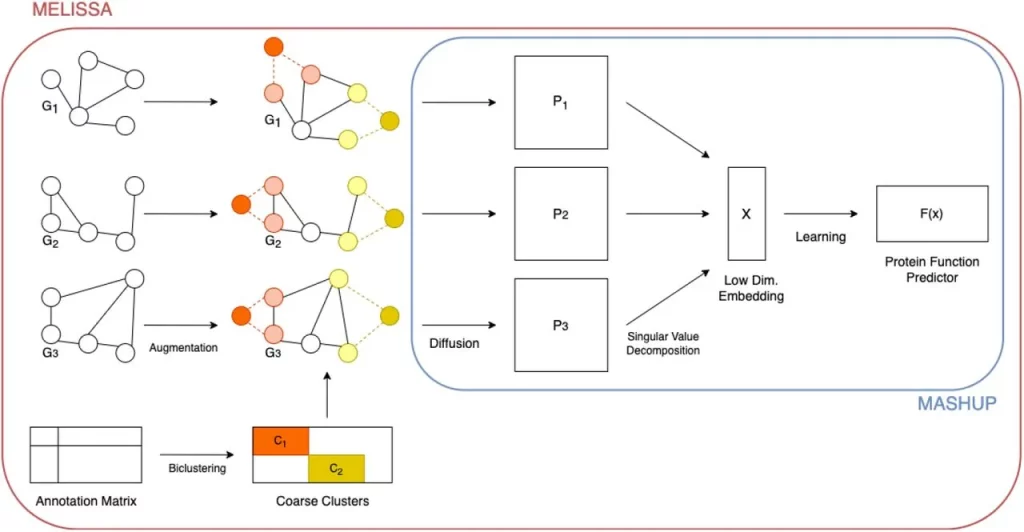
Image Source: https://doi.org/10.1101/2023.08.09.552672
Differences Among the Methodologies and Features of MELISSA, Mashup, and deepNF
MELISSA:
- Graph Augmentation: MELISSA introduces a novel step of graph augmentation by adding auxiliary nodes based on the biclustering of functional annotations. These auxiliary nodes connect to existing gene nodes and serve as anchors for functional information.
- Constraint-based Approach: MELISSA adds “must-link” and “cannot-link” constraints to the augmented graphs. These constraints guide the embedding process to incorporate functional relationships while maintaining network structure.
- Integration of Network and Functional Information: MELISSA combines network topology and functional annotations early in the process through graph augmentation and constraints. This integration aims to improve gene function prediction.
Mashup:
- Diffusion and Embedding: Mashup uses a diffusion-based approach to generate matrix representations of networks. It then combines these matrix representations to create a shared embedding using techniques like Singular Value Decomposition (SVD) or dictionary learning.
- Function Prediction: Mashup’s primary contribution is in the diffusion and embedding steps, which focus on generating a meaningful shared embedding from the matrix representations of different networks. It uses a support vector machine (SVM) or other function prediction methods to predict gene functions using the embeddings.
deepNF:
- Deep Autoencoder for Multimodal Networks: deepNF employs a deep autoencoder, specifically a Multimodal Deep Autoencoder (MDA), to generate low-dimensional embeddings of proteins across multiple networks. The method captures non-linear network structures.
- Pre-processing and Learning: deepNF involves pre-processing networks using Random Walk with Restart (RWR) to create Positive Pointwise Mutual Information (PPMI) matrices. These matrices are then used as inputs to the MDA. The middle layer of the MDA serves as the low-dimensional embedding.
- Focus on Multimodal Network Structure: deepNF’s strength lies in its ability to capture non-linear network structures across different networks using a deep neural network architecture. It focuses on learning embeddings that preserve these structures.
Experimental Evaluation of MELISSA
To assess the performance of MELISSA, the researchers conducted an experimental setup and evaluation procedure and compared it with the original Mashup and deepNF. The experiment aimed to predict Gene Ontology (GO) functional labels within biological networks for both human and yeast species. The researchers replicated the experimental configuration of the original Mashup paper to ensure consistency.
The networks used were derived from the STRING database v9.1, encompassing six heterogeneous networks for each species. While the human GO annotations remained consistent with the original Mashup, the yeast annotations shifted from the deprecated MIPS to GO annotations obtained from the Gene Ontology Consortium. Annotation specificity was addressed by retaining intermediate GO terms that annotated between 10 and 300 genes. These annotations were categorized into three functional hierarchies: Biological Process (BP), Molecular Function (MF), and Cellular Component (CC).
The evaluation process involved 5-fold cross-validation, wherein the predictive performance of the methods was assessed on both human and yeast datasets. To achieve this, the computationally efficient 𝑘-nearest neighbors (𝑘NN) method was employed for function prediction, focusing on the embedding’s influence. For each gene in the test set, its 𝑘 nearest neighbors were identified, and through a weighted majority vote accounting for pairwise distances, functional labels were predicted. Evaluation metrics encompassed percentage accuracy, F1 score, and the area under the precision-recall curve (AUPRC), with analyses conducted across different hierarchies and specificity levels of GO terms.
Observations Regarding MELISSA from the Study
This study yielded a variety of insights about MELISSA, which are elaborated upon below:
- In the provided experimental context, MELISSA consistently outperforms both Mashup and deepNF in terms of accuracy, F1 score, and AUPRC score, thereby offering a more precise representation of biological networks through its approach to function prediction.
- A range of parameter choices, particularly in yeast networks, seem to instill relative robustness to MELISSA. This suggests that the specific configurations of parameters do not overly sensitize MELISSA’s approach, thus facilitating better results without necessitating meticulous fine-tuning.
- MELISSA successfully leverages both machine learning (ML) and constraint learning (CL) constraints for enhanced function prediction, demonstrating its effective utilization of these restrictions.
- The study reveals that MELISSA performs optimally with a reduced number of biclusters (eight or fewer). Including CL constraints appears to enhance performance when dealing with a few biclusters; however, it may undermine performance in instances where the number of biclusters increases significantly.
- The superior robustness of MELISSA in yeast networks, compared to human networks, results from the availability of more functional label information for yeast. This suggests that the optimal set of parameters can vary across different GO hierarchies and levels of functional specificity, thus outlining species-dependent robustness.
Conclusion
In this study, MELISSA emerged as a better protein function predictor as compared to Mashup and deepNF. However, there is still great scope for improvement. One suggested direction is to integrate annotation information more seamlessly into the entire process, including the diffusion process (a concept likely related to how information spreads or propagates through the network). This means leveraging all available information from the outset to generate more comprehensive embeddings. Another way is by modifying network topology using constraints, such as preventing direct transitions between nodes with different functional labels. Additionally, ideas like sparsifying networks or introducing new edges are also feasible as avenues for further investigation.
Article Source: Reference Paper | MELISSA is available on GitHub
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Neegar is a consulting scientific content writing intern at CBIRT. She's a final-year student pursuing a B.Tech in Biotechnology at Odisha University of Technology and Research. Neegar's enthusiasm is sparked by the dynamic and interdisciplinary aspects of bioinformatics. She possesses a remarkable ability to elucidate intricate concepts using accessible language. Consequently, she aspires to amalgamate her proficiency in bioinformatics with her passion for writing, aiming to convey pioneering breakthroughs and innovations in the field of bioinformatics in a comprehensible manner to a wide audience.











{kind=link}