
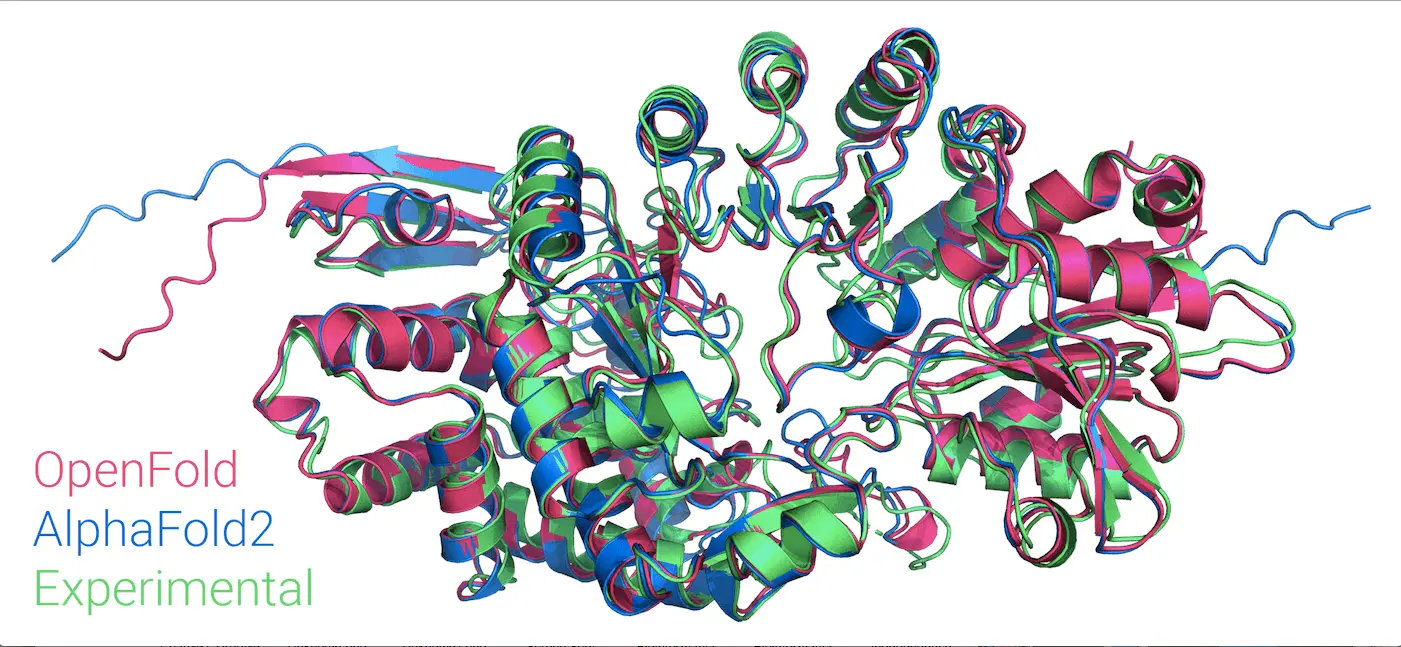
Columbia University and Harvard University researchers have developed OpenFold—a swift, memory-efficient, trainable AlphaFold2 implementation. They aimed to address the shortcomings of the revolutionary AlphaFold2, which, despite predicting protein structures with remarkable accuracy and dramatically advancing structural biology, lacked accessible code and training data. This lack limits progress in various areas, such as tackling new tasks like protein-ligand complex prediction, understanding the model’s learning process, or evaluating its adaptability to unexplored fold space. OpenFold, when trained from scratch, matches the accuracy of AlphaFold2. We assess OpenFold’s ability to generalize across fold space by retraining it with thoughtfully curated datasets; this approach reveals its robustness—impressive even in the face of significantly reduced data diversity. Moreover, our examination of OpenFold’s learning process sheds light on how it sequentially acquires spatial dimensions during protein folding. OpenFold emerges, bridging gaps for researchers in protein structure prediction and enabling further advancements in the field as a vital resource.
Why OpenFold had to be Born from AlphaFold2
For decades, deciphering protein structures from sequences has been a pivotal challenge in biology. Building upon prior deep learning work utilizing co-evolutionary data from multiple sequence alignments (MSAs) and homologous structures, AlphaFold2 has made remarkable strides in solving this problem for natural proteins with extensive MSAs. While its availability as an open-source implementation has catalyzed optimization and innovative applications, AlphaFold2’s training intricacies and resource-intensive data have been withheld, limiting deeper exploration and adaptability.
To fill this gap, researcher Mohammed AlQuraishi and his team introduced OpenFold, an open-source and trainable rendition of AlphaFold2. Developed from scratch using the OpenProteinSet, a reproduction of AlphaFold2’s training set, OpenFold mirrors AlphaFold2’s predictive quality but brings forth key advantages—enhanced speed, reduced memory usage, and compatibility with PyTorch. This framework empowers a wider community of developers and interfaces effectively with established machine learning software.
Taking advantage of their discovery that approximately 90% of model accuracy can be achieved within about 3% of the training duration, they conducted multiple retraining iterations of OpenFold using meticulously pruned training set variants. This approach aimed to systematically assess OpenFold’s capability to extrapolate to unfamiliar protein folds. Surprisingly, OpenFold demonstrated remarkable robustness even with substantial omissions of fold space, albeit with variable performance contingent on spatial extents. Intriguingly, heightened performance emerged when training on smaller yet diverse datasets, including those with as few as 1,000 experimental structures.
Further leveraging OpenFold’s capabilities, they delved into its protein-folding learning process, focusing on intermediate stages and identifying distinct phases of behavior. Through meticulous analysis of predicted structures, they unveiled OpenFold’s sequential acquisition of spatial dimensions, secondary structure elements, and tertiary scales. These revelations offer profound insights into the learning behavior of AlphaFold2-type models, equipping researchers with new conceptual and practical tools for advancing biomolecular modeling algorithms. By bridging the gap between access and understanding, OpenFold presents a pivotal resource poised to invigorate protein structure prediction research and innovation.
The Many Marvels of OpenFold
- OpenFold’s newly trained model achieves accuracy on par with AlphaFold2
OpenFold faithfully reproduces AlphaFold2’s architecture, allowing a seamless interchange of model parameters. Training is validated using diverse MSAs and structures, substituting unavailable AlphaFold2 data with OpenProteinSet. A set of 10 distinct models is generated, showing high concordance with AlphaFold2 in prediction quality. OpenFold remarkably reaches about 90% of final accuracy within 3% of training time, indicating potential for efficient exploration of model architectures. Fine-tuning, crucial for chemical constraint resolution, displays minimal impact on prediction quality, enabling swift elided training runs. Early in training, OpenFold’s self-assessment metric, pLDDT, aligns with true quality, revealing the model’s capacity for self-evaluation even during limited prediction capabilities.
- OpenFold demonstrates great accuracy even with small training sets
By training with progressively fewer protein chains, OpenFold’s accuracy (lDDT-C) is assessed. Surprisingly, even with only about 1% of the original training data, OpenFold achieves high initial accuracy comparable to the full dataset-trained model. Decreased accuracy in smaller sets is observed, but all models, including the smallest 1,000-chain sample, show notable performance. Templates’ contribution to prediction quality is limited, consistent with earlier AlphaFold2 findings.
- OpenFold shows superior generalization abilities
OpenFold’s generalization across unexplored fold space is examined through stratified training set subsampling. Entire regions are excluded in a structurally controlled manner, evaluating OpenFold’s capacity to extrapolate to out-of-distribution data. Stratifications are performed at various CATH hierarchy levels, assessing accuracy as training progresses. Despite severe reductions in diversity, even the most limited training sets achieve unexpectedly high accuracy, comparable to the initial AlphaFold model trained on substantially more data. Models trained exclusively on specific structural elements (alpha helices or beta sheets) still attain remarkable accuracy on domains containing both. This showcases OpenFold’s remarkable generalization capability.
OpenFold’s remarkable generalization capacity across excluded fold space suggests its focus on local MSA/sequence-structure correlations rather than global fold patterns. This prompts the exploration of whether its generalization is scale-dependent. By evaluating accuracy on increasing-length protein fragments using GDT_TS and lDDT-C metrics, it’s revealed that larger fragments and whole domains benefit more from increased training data. This supports the hypothesis that OpenFold’s ability to generalize relies on prediction task scale—local structures can be robustly learned even with minimal data, while global structures depend on comprehensive fold diversity representation in the training set.
- OpenFold exceeds AlphaFold2 in terms of efficiency and stability
Greater efficiency and improved stability are distinguishing features of OpenFold compared to AlphaFold2. While maintaining alignment with AlphaFold2’s computational logic, OpenFold introduces optimizations to enhance usability and performance. These optimizations include a low-precision training mode suitable for commercial GPUs, a revised approach to primary structural loss (FAPE) for better convergence, and memory-efficient training and inference stage strategies. These adaptations result in significantly enhanced efficiency, making OpenFold up to three times faster in inference for shorter proteins and capable of processing longer proteins and complexes without memory constraints. Additionally, OpenFold’s training speed matches or surpasses that of AlphaFold2, as observed in various research studies.
OpenFold’s Learning Process: The Secret to Its Success
OpenFold’s learning process sheds light on its mastery of protein structure prediction. During the initial 5,000 training steps, the model’s progression unfolds in a staggered manner, focusing on local patterns before the global structure. Predictions evolve from point-like to one-dimensional structures, gradually gaining orthogonal dimensions, forming secondary structure elements, inflating to attain backbone structures, and refining atomic details.
The sequential progression suggests OpenFold acquires spatial dimensions sequentially, learning a series of lower-dimensional PCA projections of the final 3D structures. Analysis substantiates this by comparing PCA projections to the 3D prediction, highlighting convergence over dimensions. Notably, OpenFold’s capacity for such learning is attributed to its freedom from strict physical priors, enabling low-dimensional structures with steric clashes and chemical law violations—a crucial ingredient in its success.
OpenFold follows a staggered and multi-scale pattern in learning secondary structure elements (SSEs). Analysis of SSE F1 scores during training reveals that alpha helices are learned first, followed by beta sheets, and then less common SSEs, corresponding to their prevalence in real proteins. This sequence is consistent with the order of SSE frequency in proteins, with a slight exception for rare helix variants. The recognition of SSEs lags behind the accuracy of global structural predictions. In the learning of helices, it is found that high F1 scores for alpha helices are achieved by OpenFold all at once, not gradually through fragments.
Similarly, in terms of tertiary structure, local fragments are initially excelled at being predicted by OpenFold before global structures are refined. This trend is also observed when assessing secondary structure elements, with short helices and narrow sheets showing better prediction early in training. This multi-scale learning behavior eventually leads to a scale-independent predictive capacity, as evidenced by the consistent accuracy achieved for various SSE lengths and widths. The ability of the model to capture these features may also extend to aspects unrecognized by traditional structural analysis methods like DSSP.
Conclusion
Researchers built OpenFold to reproduce the protein structure prediction capabilities of AlphaFold2. The successful training of OpenFold from scratch and its achievement in matching the accuracy of AlphaFold2 confirm the robustness of this model for predicting protein structures. In addition, Openfold advances technically by notably enhancing prediction speed and utilizing PyTorch, a prevalent deep-learning framework. This implementation enables deeper analyses of AlphaFold2-like models and supports the development of new (bio)molecular models leveraging AlphaFold2 modules.
OpenFold demonstrates a capacity, as highlighted in the study, to learn from reduced training sets and reveal the sequential learning behavior of spatial dimensions prevalent in models such as AlphaFold2. These insights potentially inform how we can incorporate physical priors into machine-learning models and present a curriculum-learning approach for problem-solving.
Article Source: Reference Paper | OpenFold can be accessed at GitHub | Featured Image Source: GitHub
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Neegar is a consulting scientific content writing intern at CBIRT. She's a final-year student pursuing a B.Tech in Biotechnology at Odisha University of Technology and Research. Neegar's enthusiasm is sparked by the dynamic and interdisciplinary aspects of bioinformatics. She possesses a remarkable ability to elucidate intricate concepts using accessible language. Consequently, she aspires to amalgamate her proficiency in bioinformatics with her passion for writing, aiming to convey pioneering breakthroughs and innovations in the field of bioinformatics in a comprehensible manner to a wide audience.











{kind=link}