
Accurately identifying somatic mutations and differentiating them from germline variations is critical for precision oncology and precise tumor-mutational burden (TMB) computation. For this purpose, three trained machine-learning models, TabNet, XGBoost, and LightGBM, were utilized, and state-of-the-art performance was achieved, with concordance between matched-normal and tumor-only TMB improving from R2=0.006 to R2=0.73. The racial biases in germline databases are removed, to some degree, using different techniques such as LightGBM and XGBoost.
Importance of accurate estimation of Tumour-mutational burden
Tumor mutation burden (TMB) is a powerful predictor of therapeutic response and survival in solid tumors. TMB determines the number of coding nonsynonymous somatic mutations per megabase of DNA. Along with TMB, it is critical to characterize somatic mutations and understand germline variations to forecast whether a cancer patient will react to current therapies. The lack of a patient-matched normal hinders somatic variant identification in precision oncology, resulting in a 67% false positive rate. This implies that most putative somatic mutations in tumor-only variant calling are simply rare germline variations. As a result, tumor-only TMB is inflated compared to “true” TMB calculated by germline variant removal using a matched-normal. The complexity of the cancer genome, including clonality and structural variation, combined with the detailed statistics of next-generation sequencing, makes it difficult to improve upon these statistical models. Several computational methods have been developed to enhance tumor-only variant calling. XGBoost and LightGBM, tree-based ML algorithms with gradient boosting, and TabNet, a deep-learning approach for tabular features, are used to distinguish somatic and germline variations in cancer data. Additionally, ML classifiers can completely eliminate racial bias brought on by the underrepresentation of minorities
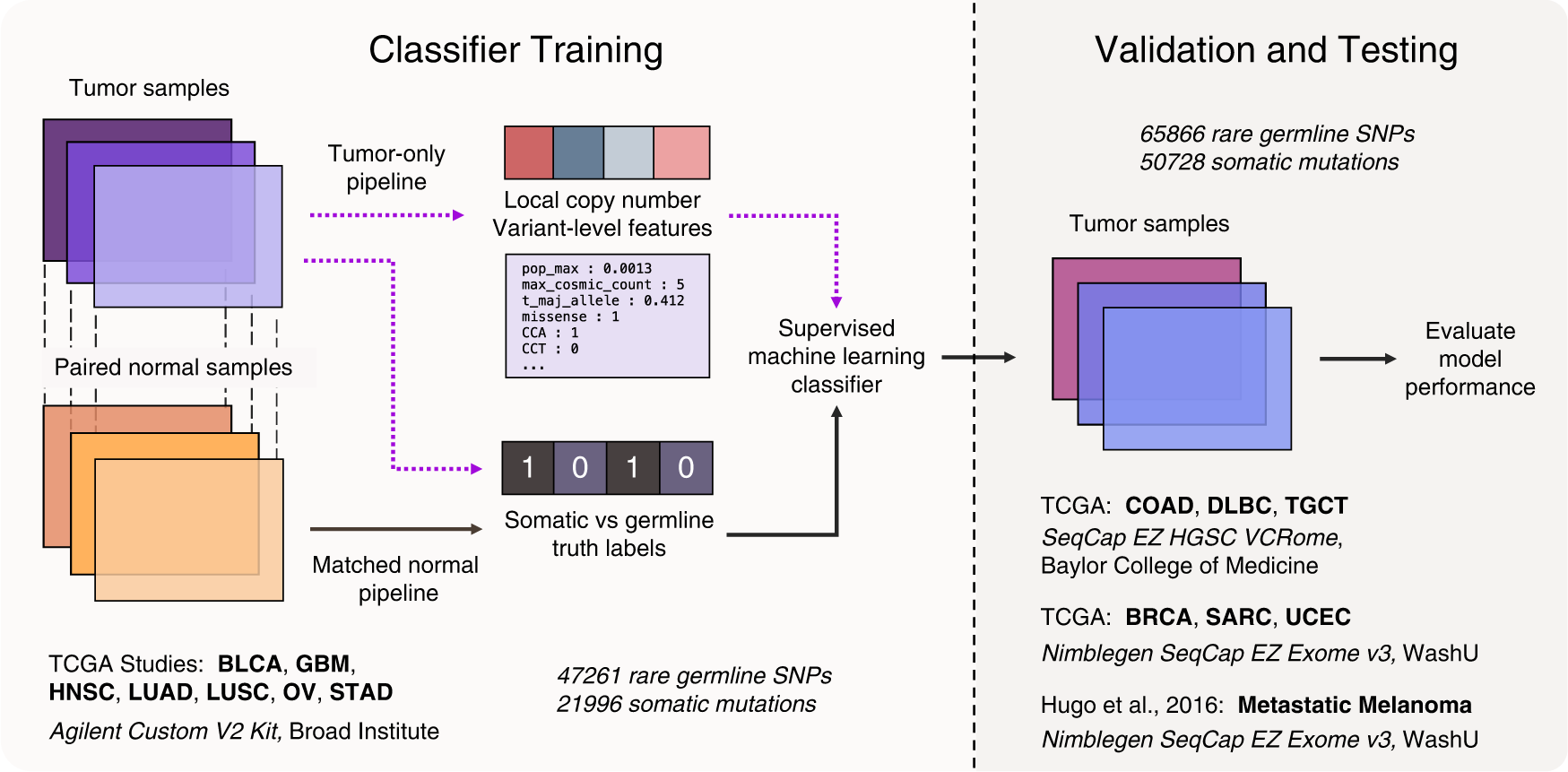
As TMB, copy-number variation, and tumor purity may all impact somatic mutation calling, oncology samples that ranged in biological extremes from testicular germ cell cancer (very low TMB) to squamous carcinoma (high TMB) were employed for the training and testing of the ML models.
Building the Machine-Learning models
Using tumor-only samples, 30 mutation- and copy-number-specific features were created. This included conventional features for somatic variant calling, such as germline database frequency, COSMIC somatic mutation database counts, and major allele frequency.
An independent variant-calling pipeline was used to determine the somatic and germline truth labels for the ML models using the matched-normal samples. Mutations passing via the matched normal pipeline were classified somatic, while all other variants passing through the tumor-only route were considered germline.
There were 105 tumor samples chosen from different patients in seven cancer subtypes from The Cancer Genome Atlas (TCGA) for our training set. The findings of a variant-calling process that included patient-matched-normal samples were used to establish somatic and germline truth labels. The validation set included 45 tumor samples from three cancer subtypes: colon adenocarcinoma (COAD), lymphoid neoplasm diffuse large B-cell lymphoma (DLBC), and testicular germ cell cancers (TGCT). These, however, were not included in the training set. The optimally trained models have AUCs (Area under the ROC curve) of 0.96 (TabNet), 0.98 (LightGBM), and 0.99 (XGBoost) for fitting the training data. An ensemble average was also created, which is a simple average of the posterior probabilities of the three models for better classification.
Following model training and selection, two different holdout test sets were made with four cancer subtypes. These findings show that the biological differences between cancer tissue subtypes have a greater impact on performance than the machine-learning model used. A final holdout dataset was obtained to evaluate further the models’ generalization and resistance to batch effects. LightGM outperformed the other models in AUC, Matthews Correlation Coefficient (MCC), and balanced accuracy.
How the ML models improved TMB estimation
Reliable estimation of TMB is an essential criterion for a model developed to enhance tumor-only variant calling. This capability is instrumental in immuno-oncology clinical trials, where TMB is a powerful indicator of response and survival, and matched-normal samples are not always available. Compared to the naive technique, which is a non-machine-learning naive tumor-only strategy that includes a process-matched panel of normals, various germline variant databases, and conventional variant filtering approaches to eliminate germline variations and artifacts, the machine-learning model achieves a 45-50-fold improvement. These tabular models’ improvement argues for adopting ML-adjusted TMB estimations for clinical variant analysis in tumor-only samples.
Eliminating racial bias from the TMB estimations
The underrepresentation of racial minorities in genomic databases has significant repercussions in human genome science and has been heavily criticized. Tumor-only variant calling is no different. According to a recent study, the inflating of TMB induced by the lack of a matched-normal sample is especially severe among underrepresented minorities. After using the tabular model predictions to exclude uncommon germline variations, the inflation of TMB for Black patients is considerably decreased and similar to that of white patients. The p-value, which indicates how likely it is for a hypothesis to be true, is not significant for XGBoost and LightGBM, suggesting that bias has been completely eliminated.
Improvements over PureCN
PureCN is an open-source R package used to detect copy number variation and single nucleotide variation classification in targeted sequencing data.
There is a significant increase in computation speed over PureCN, which is not surprising given that trained, supervised machine-learning classifiers have a lower CPU need than Bayesian approaches. Tabular ML models outperformed linear ML models overall and on SNVs in the holdout datasets.
The absence of racially biased TMB inflation in these models is likely due to the lesser reliance on population datasets. Perhaps understanding count, the total number of mutations to classify—which is heavily influenced by the number of uncommon germline variations missing from biased databases—allows LightGBM and XGBoost to detect and make better judgments with samples from underrepresented populations.
Using multiple regression models, individual performance is explained as a function of various predictor factors. The “true” TMB obtained through germline subtraction via the matched-normal pipeline was found to be the most impactful element on a sample’s positive predictive value (PPV). Samples having a lower TMB have a lower PPV. Despite being highly predictive of model success, the genuine TMB can only be computed in the absence of a matched-normal sample.
Three types of tabular machine learning (ML) classifiers were built and trained to differentiate somatic mutations from uncommon germline variations. The models were trained on seven cancer subtypes that were sequenced using a single exome capture kit. In terms of speed and overall accuracy, tabular ML models surpass PureCN.
Concluding remarks
Despite TabNet’s promising claims, the tree-based XGBoost and LightGBM approach somewhat outperformed it. However, adding TabNet improved the average ensemble predictions, indicating that the single optimal model depends on the classification instance. The fact that not all subpopulations are well-represented in genetic research is a major constraint in human genomics and precision medicine. The algorithms used were able to significantly reduce the racially biased overestimation of tumor-only TMB with TabNet and PureCN, and eliminate this bias below the practical limit, by integrating multiple informative features such as the total number of variants to classify (somatic + rare germline SNPs), COSMIC and germline databases, and variant allele fractions.
Article Source: Reference Paper
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Sejal is a consulting scientific writing intern at CBIRT. She is an undergraduate student of the Department of Biotechnology at the Indian Institute of Technology, Kharagpur. She is an avid reader, and her logical and analytical skills are an asset to any research organization.





{kind=link}