

Computational tools that are currently used for molecular docking are difficult to integrate into different computational distributions and interfaces; this becomes very inconvenient for users and researchers. EasyDock is a novel tool that can be applied to a network of computational nodes aided by the Dask Library and does not require scheduling for any specific clusters of compounds. The researchers involved in the development of EasyDock implemented a model that can predict the time that will be taken by docking experiments and can simultaneously reduce overall docking time. EasyDock currently supports docking programs that are commonly used, such as smina, gnina, and AutoDock Vina. Some additional features include a supplementary feature that facilitates the docking of compounds that contain boron and 55 other protein-ligand complexes extracted from the Protein Data Bank (PDB).
An introduction to molecular docking
Ideally, computational tools used for molecular docking should be able to easily integrate into existing interfaces and be easy to navigate for users. The convenience of usage would encourage a greater number of researchers to carry out docking procedures for their experiments. The discovery of novel chemical compounds is made possible through the process of docking across large collections of compounds. De novo design projects that involve molecular docking also require the screening of compounds on a large scale. These processes require automatic tools that are capable of docking molecules with the help of computational nodes in a short period of time.
In the early stages of drug discovery, the first step is to identify promising hits in the chemical landscape. This is achieved by high-throughput screening (HTS), which screens chemical libraries containing millions of compounds and identifies initial hits. These seemingly infinite chemical libraries constitute only a small portion of the drug-like chemical space in its entirety, which is estimated to have nearly 1036 compounds contained within it. This number has been further increased with the addition of combinatorial libraries encoded with data based on DNA, due to which even a single docking campaign will cover compounds in a range of 109 to 1010. A downside here, however, is that DNA-encoded libraries are limited as not all chemical library reactions are suited for coupling building blocks; therefore, the chemical space is not covered entirely. This is solved using improved and more advanced computational approaches.
Computational approaches to docking
Computational approaches can be classified into two types: de novo designs and virtual screenings. De novo design (DND) of drugs involves designing novel chemical compounds that fit into a set of criteria with the use of computational growth algorithms. To obtain hits that are highly active, virtual screening of ultra-large libraries has proved to be very effective. However, its application remains limited due to many technical difficulties.
In place of virtual screening, de novo design (DND) approaches give much more room for exploration. Within the DND domain, the iterative generation of molecules takes place according to certain criteria. This is referred to as the docking score. It helps in exploiting only certain regions within the chemical space that contain promising hits rather than scaling it in its entirety. Such methods have exhibited high efficacy in discovering hits, with the number of docked molecules reaching up to millions. Whether researchers choose DND or virtual screening as their method of choice, the final goal is to dock as many molecules as possible within a single campaign.
A Python module called DockString was developed previously to tackle the limitations posed by other docking tools when it comes to integration into different systems. However, users can only use this module as a basis for creating their own distributed workflow.
Introducing EasyDock
EasyDock can undertake calculations for either a single or several servers located within a given network. It can be obtained either from command lines or in the form of a Python module; this expands its applications for molecular docking.
A notable feature of EasyDock is its ability to dock compounds containing boron. Functional groups containing boron have the ability to form strong hydrogen and covalent bonds, regulate pharmacokinetic activities, and offer resistance to drugs. For this reason, scientists working on drug discovery are interested in them. Despite all the potential held by compounds containing boron, most of the commonly used docking programs don’t support them.
Researchers took the approach of replacing boron with carbon, as it has been hypothesized that both of them have similar atomic properties. They ran docking processes on the compounds afterward. The boron functional groups would be put back into their places after docking was over. The output of EasyDock is just a single database from which the data can be conveniently extracted using SQL queries. EasyDock can be implemented using CPUs and even GPUs if the integrated docking programs support it.
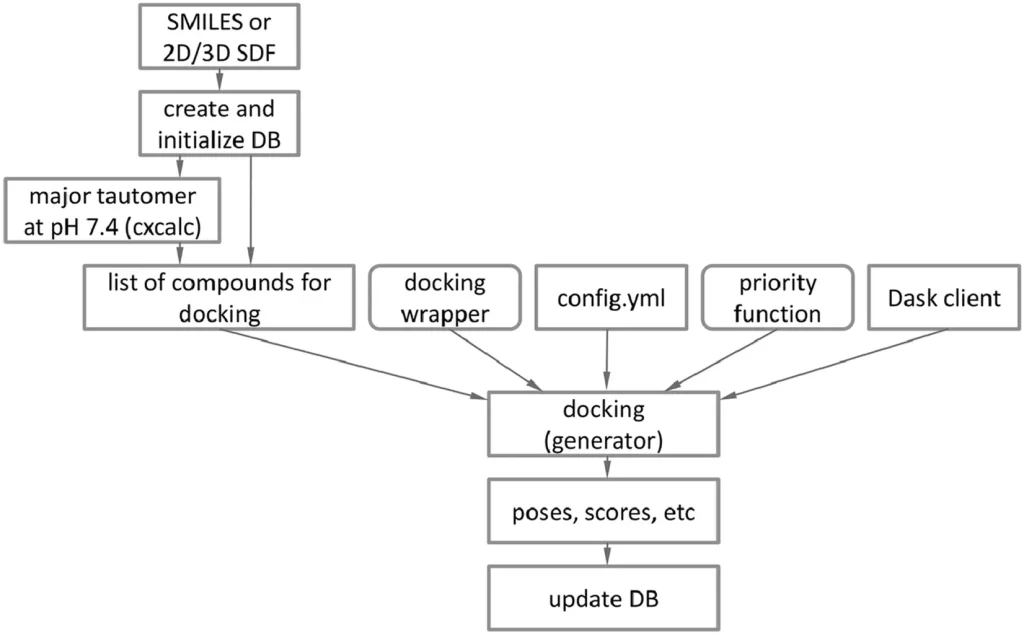
Image Source: https://doi.org/10.1186/s13321-023-00772-2
Comparison of EasyDock with other programs
AutoDock Vina is one of the most integrative docking programs currently used. EasyDock offers everything AutoDock Vina does while extending its applications to include computationally heavy programs such as gnina. This gives it an advantage over other programs because gnina makes use of 3D convolutional neural networks.
Tautomers are isomeric compounds that can readily interconvert, while stereoisomers are compounds that have the same molecular formula but spatially different chemical structures. When discussing the generation of these compounds, EasyDock is capable of generating both of them automatically. The Chemaxon cxcalc utility in EasyDock can also protonate the tautomers. Existing programs like DockString and VirtualFlow lack the capacity to generate tautomers and stereoisomers, respectively.
A Python API (application programming interface) provided by EasyDock enables its seamless integration into third-party software. It does not rely on any scheduler to facilitate distributed computing over various network devices. Commonly used schedulers such as PBS and SLURM do not provide any API for integration into different software.
Conclusion
EasyDock is designed to reduce user intervention in docking and returns the output to all given inputs within a single database. It also operates a linear model to select compounds in order of priority for docking and is readily extendable to other programs. It is open source and easily accessible to everyone in the scientific community. Upcoming improvements to EasyDock will involve scalability testing and adding a function to count the number of stereoisomers it produces.
Article source: Reference Paper | Project home page: GitHub
Learn More:




Swasti is a scientific writing intern at CBIRT with a passion for research and development. She is pursuing BTech in Biotechnology from Vellore Institute of Technology, Vellore. Her interests deeply lie in exploring the rapidly growing and integrated sectors of bioinformatics, cancer informatics, and computational biology, with a special emphasis on cancer biology and immunological studies. She aims to introduce and invest the readers of her articles to the exciting developments bioinformatics has to offer in biological research today.





{kind=link}