

Scientists from the California Institute of Technology, United States, have developed a python package, ‘BioCRNpyler’, to build, manage, and explore intricate biochemical models. BioCRNpyler can be used to speed up the computer-assisted design of biochemical circuits and the creation of model-driven hypotheses in biology.
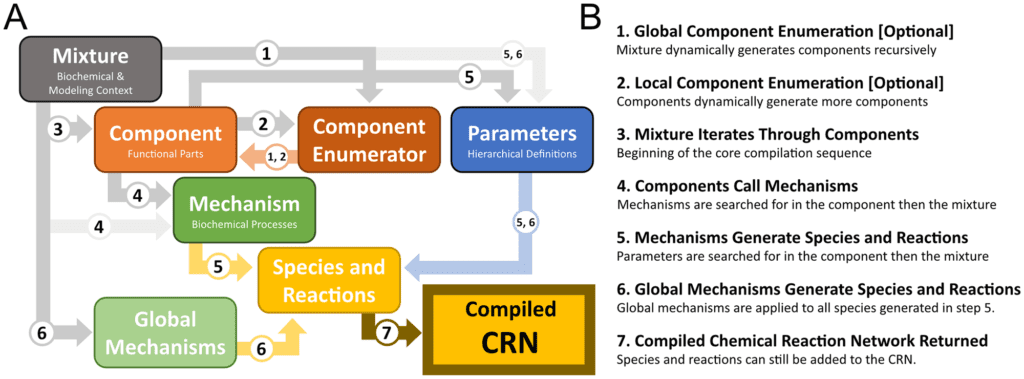
Image Source: BioCRNpyler: Compiling chemical reaction networks from biomolecular parts in diverse contexts
Biochemical interactions in systems and synthetic biology are frequently modeled with chemical reaction networks (CRNs). CRNs give a principled modeling environment prepared to express a gigantic range of biochemical cycles. In this paper, the scientists present a software toolbox written in Python that compiles high-level design specifications represented by utilizing a modular library of biochemical parts, mechanisms, and contexts for CRN executions. This compilation process offers four benefits:
In the first place, the building of the actual CRN representation is automatic and yields Systems Biology Markup Language (SBML) models compatible with various simulators as output.
Second, a library of modular biochemical components considers various architectures and implementations of biochemical circuits to be addressed briefly, with design choices spread all through the underlying CRN automatically. This precludes the frequently happening mismatch between high-level design and model dynamics.
Third, high-level design specifications can be embedded into different biomolecular environments, like without cell extract and in vivo milieus.
At last, the software toolbox has a parameter data set, which permits users to rapidly prototype huge models utilizing a few parameters which can be customized later. By utilizing BioCRNpyler, users ranging from expert modelers to beginner script-writers can undoubtedly build, manage, and explore intricate biochemical models using diverse biochemical implementations, environments, and modeling presumptions.
The Chemical Reaction Networks or CRNs
Chemical reaction networks (CRNs) are the workhorse for modeling in systems as well as synthetic biology. The strength of CRNs lies in their expressivity; CRN models can go from physically realistic depictions of individual molecules to coarse-grained idealities of complex multi-step processes.
In any case, this expressivity includes some major disadvantages. Picking the right level of detail in a model is more an art than a science. The modeling system requires careful consideration of the desired utilization of the model, the available information to parameterize the model, and the prioritization of specific aspects of modeling or analysis over others.
Moreover, biological CRN models can be unbelievably complex, including dozens or even hundreds of thousands of species, reactions, and parameters. Keeping up with complex hand-built models is challenging, and errors can rapidly grow out of control for enormous models. Software tools can solve many of these difficulties by automating and streamlining the model construction process.
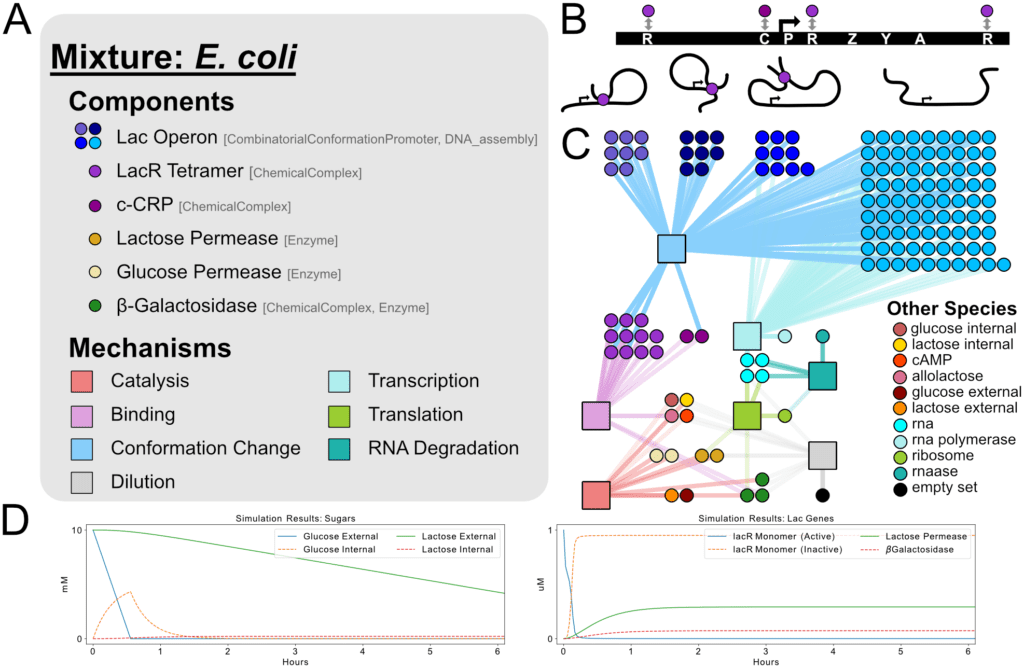
Image Source: BioCRNpyler: Compiling chemical reaction networks from biomolecular parts in diverse contexts
The BioCRNpyler
As the name would propose, BioCRNpyler (pronounced as bio-compiler) is a Python package that compiles CRNs from the exact specifications of biological motifs and contexts. This package is inspired by the molecular compilers created by the DNA-strand displacement community and molecular programming communities, which, overall, aim to compile models of DNA circuit implementations from less complex CRN specifications, rudimentary programming languages, and abstract sequence specifications.
This body of work has exhibited the utility of molecular circuit compilers and highlights that a single specification can be compiled into numerous molecular implementations, which can thus correspond to different CRN models at different levels of detail. For instance, there are multiple DNA-strand implementations of catalysis, and the interactions of the DNA strands engaged with every one of these implementations can be counted to generate unique CRN models in light of the assumptions underlying the enumeration algorithm.
Drawing from these motivations, BioCRNpyler is a general-purpose CRN compiler equipped for converting abstract specifications of biomolecular parts into CRN models with full programmatic command over the compilation process. Critically, BioCRNpyler isn’t a CRN simulator—models are saved in the Systems Biology Markup Language (SBML) to be viable with the user’s simulator of choice.
BioCRNpyler complements existing software packages by giving a novel abstraction and framework which takes into consideration complex CRNs to be easily generated and explored using the compilation process.
To do this, BioCRNpyler specifies a biochemical framework as a set of modular biological parts, biochemical processes codified as CRNs, and biochemical and modeling context.
Besides, BioCRNpyler takes into consideration synthetic biological parts and systems biology motifs to be reused and recombined in diverse biochemical contexts at customizable levels of model complexity with negligible coding prerequisites (BioCRNpyler is intended to be a scripting language).
Also, BioCRNpyler is intentionally suited to in silico work processes since it is an extendable object-oriented framework composed entirely in Python that incorporates existing software development standards and permits complete oversight over model compilation.
Simultaneously, BioCRNpyler speeds up model construction with extensive libraries of biochemical parts, models, and examples pertinent to synthetic biologists, bio-engineers, and systems biologists. The BioCRNpyler package is accessible on GitHub and can be introduced using the Python package index (PyPi).
The Takeaway and Future Prospects of BioCRNpyler
BioCRNpyler expects to be a piece of open-source community-driven software that is, without any problem, open to biologists and bioengineers with varying levels of programming experience as well as effectively customizable by computational biologists and further advanced developers.
Towards these ends, the software package is accessible through GitHub and PyPi, requires exceptionally minimal software dependencies, and contains extensive examples and documentation such as interactive Jupyter notebooks, YouTube instructional videos, and automated testing to guarantee stability.
Moreover, this software has been extensively tested through incorporation in bio-modeling courses and boot camps for users ranging from college freshmen and sophomores with minimal coding experience to advanced computational biologists showing the accessibility and adaptability of the package.
BioCRNpyler has proactively been deployed to construct diverse models in synthetic biology, including modeling bacterial gene regulatory networks, modeling bacterial circuits in the gut microbiome, and modeling cell extract metabolism.
BioCRNpyler is a continuous effort that would develop and change with the necessities of its community. Broadening this community through action, documentation, and a consistently extending set-up of functionalities is vital to the objectives of this task.
The scientists were especially keen on working with the integration of BioCRNpyler into existing lab pipelines to make modeling a focal piece of the design-build-test cycle in synthetic biology.
One road towards this objective is to add compatibility to existing standards, for example, SBOL and automation platforms such as DNA-BOT, so BioCRNpyler can automatically compile models of circuits as they are being designed and built.
The scientists additionally planned on stretching out the library to incorporate more realistic and diverse mixtures, mechanisms, and components (especially experimentally validated models of circuits in E. coli and cell extracts).
They hoped those models would act as specific illustrations and motivation for different researchers to add their model frameworks in other organisms to the software library.
The researchers believed that the Mixture-Component-Mechanism abstraction of model compilation utilized in BioCRNpyler is essential and could be stretched out to other non-CRN-based modeling approaches.
Advanced simulation techniques past chemical reaction networks would be required to model the diversity and complexity of biological systems precisely. For example, in new software frameworks, Vivarium can create models that couple numerous simulation modalities.
The abstractions utilized in BioCRNpyler could be stretched out to compile models past chemical reaction networks like mechanical models, flux balance models, and factual models derived from information.
The integration of these models will typically rely upon both points by point mechanistic descriptions and overarching system context.
The scientists emphasize that building extendable and reusable systems to empower quantitative modeling in biology will become progressively essential to comprehend and design perpetually more complex biochemical frameworks.
Article Source: Poole W, Pandey A, Shur A, Tuza ZA, Murray RM (2022) BioCRNpyler: Compiling chemical reaction networks from biomolecular parts in diverse contexts. PLoS Comput Biol 18(4): e1009987. https://doi.org/10.1371/journal.pcbi.1009987
Data Availability: BioCRNpyler source code and an extensive set of example notebooks, documentation, and tutorials are available in our GitHub repository: https://github.com/BuildACell/BioCRNPyler.
Tanveen Kaur is a consulting intern at CBIRT, currently, she's pursuing post-graduation in Biotechnology from Shoolini University, Himachal Pradesh. Her interests primarily lay in researching the new advancements in the world of biotechnology and bioinformatics, having a dream of being one of the best researchers.







{kind=link}