

A team of researchers at the University of California, San Diego, came together to develop a chatbot specifically catered to drug discovery called DrugChat. It is similar to ChatGPT on the basis that users can give question prompts to the model on any drug compound under study after uploading the drug’s molecular graph and can get answers to their queries in an interactive and iterative manner. It is composed of a graph neural network (GNN), a large language model (LLM), and an adaptor.
Traditional methods of drug discovery, which have been carried out experimentally, have been expensive and time-consuming. Approval for even a single drug to go up in pharmaceutical stores for sale takes several years and a lot of money. Experimental methods also require a lot of labor and step-by-step testing, often ending up in failure at the last few stages of drug development. Taking advantage of artificial intelligence (AI) to mitigate these issues is crucial in today’s fast-paced and demanding world, where novel drugs are needed to treat an ever-increasing list of diseases.
An insight into how the model works
Using the model itself is a relatively simple process, as it can be used just like ChatGPT; all that the user needs to do here is upload the molecular graph of the compound they want to study, and they can then proceed to ask whatever questions they desire about the compound.
The model has three major components that work sequentially: a graph neural network (GNN), a large language model (LLM), and an adaptor. The user’s molecular graph serves as the input for the GNN, which learns a representation of the graph and generates a representation vector before sending it to the adaptor. The adaptor converts the data retrieved from the representation vector into a prompt vector, a representation type that the LLM can comprehend. Once the prompt vector reaches the LLM, it takes in the information given to it and uses it to answer questions asked by the user. Weight parameters transform input data present within a neural network’s hidden layers. In this model, the weight parameters remained fixed for the GNN and LLM and were variable for the adaptor.
The datasets for drug compounds were created using data from PubChem and ChEMBL. From each of these databases, compounds that were drugs were identified, and information regarding them was extracted and gathered. SMILES strings were obtained for each drug, representing their molecular structures. Information regarding the general features as well as features specific to different drugs was gathered as well.
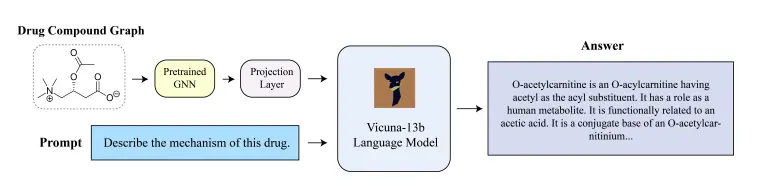
Image Source: https://doi.org/10.48550/arXiv.2309.03907
A Revolution for Pharmaceutical Research
DrugChat has the potential to revolutionize pharmaceutical research as we know it. It can reduce the risks of final-stage failures in drug discovery by allowing researchers to thoroughly analyze drugs before carrying out experimentation using AI.
Molecular graphs are difficult to understand and analyze as they differ greatly from textual data. Converting them into a form that is understandable for AI is vital, as it can help predict the properties of compounds more efficiently and can give suggestions to optimize drug design. Providing a thorough overview of the drug’s side effects and therapeutic properties beforehand can speed up the drug discovery process by reducing the time taken during the initial stages. It can also compare the molecular structures of known compounds and use this to predict the interactions between new and existing drugs, as well as the potential behavior of drugs in real-life scenarios.
Structure-activity relationship (SAR) is a very important aspect to consider when designing drugs. It gives an understanding of the relationship between the biological function of the drug and its structure. DrugChat can help understand the structure of the drug and identify any structural changes that need to be made to reduce side effects.
Lead compounds are promising drug candidates that appear during initial drug screening procedures. DrugChat can identify these compounds and suggest changes that can enhance their desirable effects and reduce their toxicity. This would save a lot of time and effort. It can also identify drugs that have the potential to treat diseases that they were not initially designed to treat. This gives a quicker method for curing challenging diseases. DrugChat can also help streamline tasks carried out during clinical trials.
Challenges in the development of DrugChat
The developers faced three major challenges:
- Molecular Graphs: They are different from the usual text-based data interpreted by LLMs. Textual data is well-defined and sequential, while molecular graphs are much more complex and unorganized. They do not have a predefined start or end point. It was important to devise a method to process these graphs into a format that could be understood by the LLM.
- Structure-Activity Relationships are complicated: SARs are not easy to understand, as these are very intricate relationships. They are not directly depicted, and many interactions can be too subtle to capture.
- Requirement of large datasets: The generation and compilation of large amounts of data is not an easy task. Here, a very diverse collection of data comes into play, and some information can also be proprietary and restricted.
Conclusion
When DrugChat was tested by developers, they found that it was able to provide intelligible and sensible responses for questions that were not present in its training dataset as well. It was able to answer a diverse array of questions, thus exhibiting the potential to enable a ‘conversational analysis’ of drug compounds.
A possible limitation of DrugChat can be language hallucinations, a recurring issue in LLM models. This can result in the model giving untrustworthy answers, potentially jeopardizing the entire clinical trial protocol. This problem can be mitigated by using higher-quality datasets and effective filtering methods, which can be made possible by incorporating more advanced GNNs and LLMs into the framework of DrugChat. Once made available to the public, widespread use of DrugChat by the public can create a human feedback mechanism, which can act as a check and balance to prevent the model from giving incorrect answers.
Therefore, DrugChat has the potential to accelerate drug discovery and can act as a valuable and useful addition to the pipeline of drug discovery and development.
Article source: Reference Paper | The code and data is available on GitHub
Important Note: arXiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Swasti is a scientific writing intern at CBIRT with a passion for research and development. She is pursuing BTech in Biotechnology from Vellore Institute of Technology, Vellore. Her interests deeply lie in exploring the rapidly growing and integrated sectors of bioinformatics, cancer informatics, and computational biology, with a special emphasis on cancer biology and immunological studies. She aims to introduce and invest the readers of her articles to the exciting developments bioinformatics has to offer in biological research today.











{kind=link}