
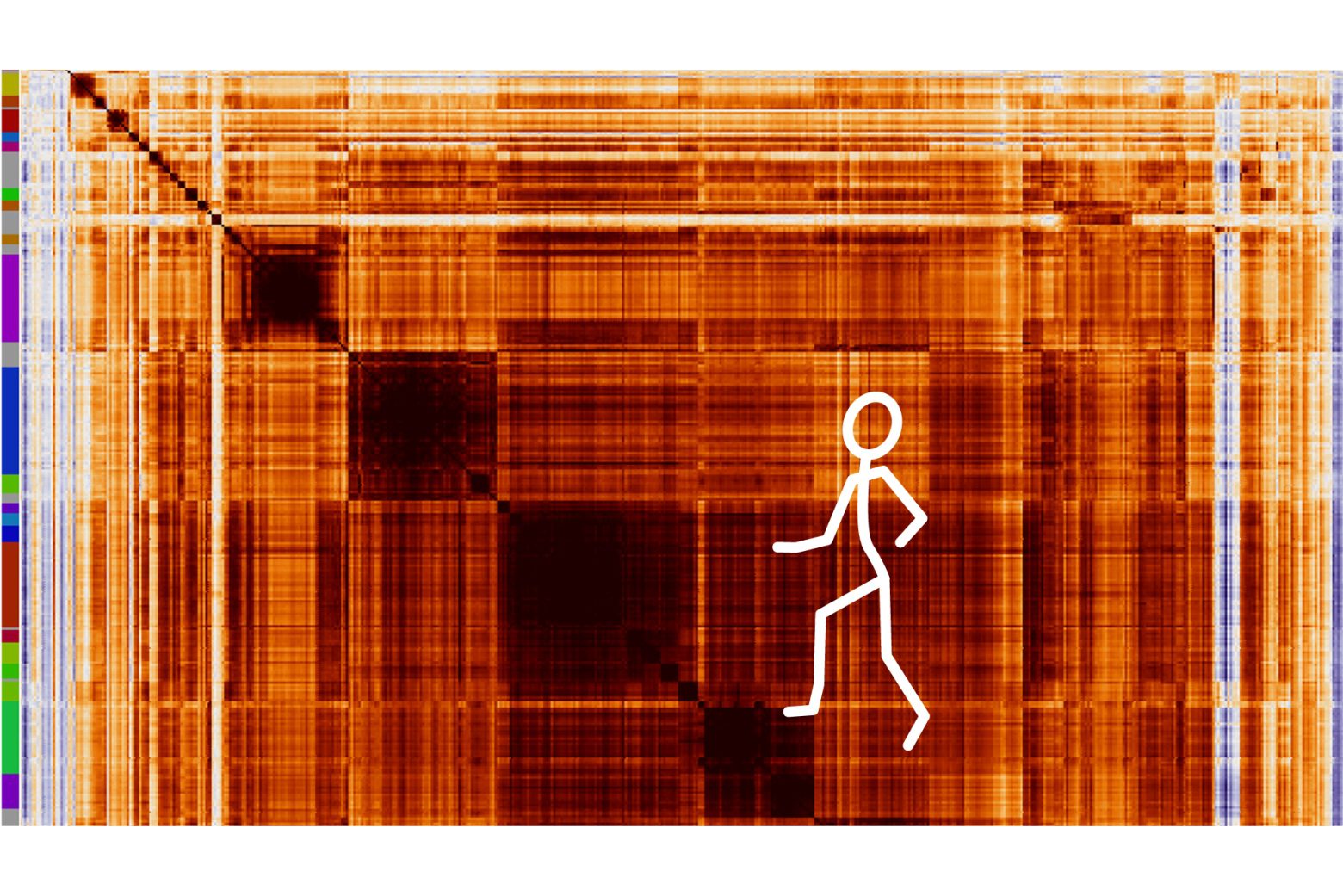
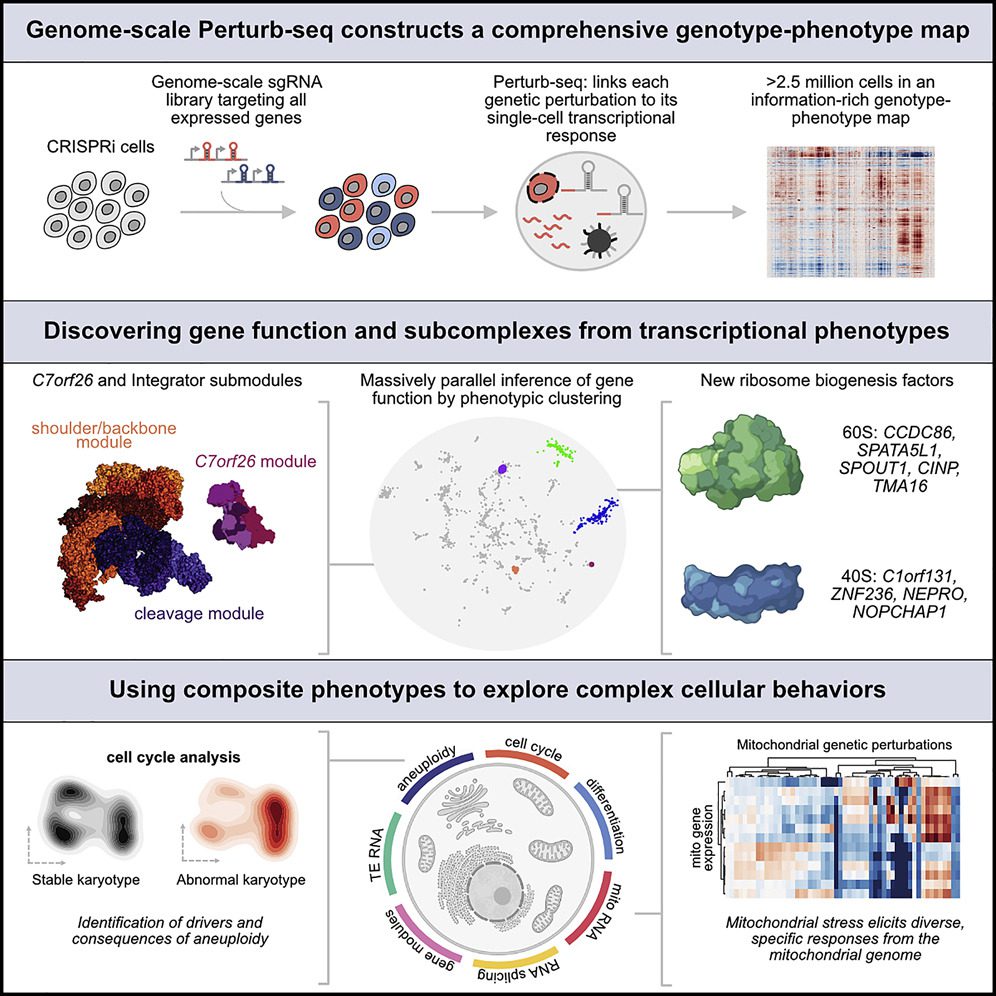
Scientists from Whitehead Institute and collaborators used Perturb-seq, a single-cell sequencing technology, to link every expressed gene in the human genome to its function in the cell, offering a free resource for other researchers that can be accessed via the internet.
The Human Genome Project was a huge undertaking that aimed to sequence every single strand of human DNA. The project, which included researchers from Whitehead Institute and other research institutes throughout the world, was finally finished in 2003.
After more than two decades, Whitehead Institute Member Jonathan Weissman and colleagues have presented the first comprehensive functional map of genes expressed in human cells. The findings, which were published online in Cell on June 9th, link each gene to its function in the cell and are the result of years of collaboration on the single-cell sequencing method Perturb-seq.
Understanding gene and cellular function require mapping the connection between genetic changes and their phenotypic consequences. A phenotype-centric, “forward genetic” technique shows the genetic alterations that drive a phenotypic of interest, while a gene-centric, “reverse genetic” approach catalogs the diverse phenotypes generated by a specific genetic change.
CRISPR tools now make it simple to delete, mutate, repress, or activate genes. CRISPR-Cas systems can be used to forward genetic screens for generating pools of cells with a variety of genetic perturbations, which can then be subjected to selection and sequencing to ascribe phenotypes to the genetic perturbations.
It’s a tremendous resource in the same sense that the human genome is a great resource in that you can go in and do discovery-based research. Rather than deciding ahead of time what biology you’re going to look at, you have this map of genotype-phenotype connections and you can go into the database and screen it without having to do any studies.
Jonathan Weissman, who is also a professor of biology at MIT and a Howard Hughes Medical Institute investigator.
The screen enables the researchers to investigate a wide range of biological issues. They utilized it to analyze the cellular impacts of genes with unclear roles, the response of mitochondria to stress, and to look for genes that induce chromosomal loss or gain, a characteristic that has hitherto been difficult to study.
I think this dataset is going to enable all kinds of analyses that we haven’t even thought of yet by people who come from different parts of biology, and suddenly they just have this ready to draw on.
Tom Norman, a former postdoc at the Weissman Lab and a co-senior author of the research.
Escalating Perturb-seq
The Perturb-seq technique is used in this investigation, which allows researchers to follow the impact of turning genes on or off in unprecedented detail. This approach, which was first described in 2016 by a group of researchers led by Weissman and fellow MIT professor Aviv Regev, could only be employed on a small number of genes and at a high cost.
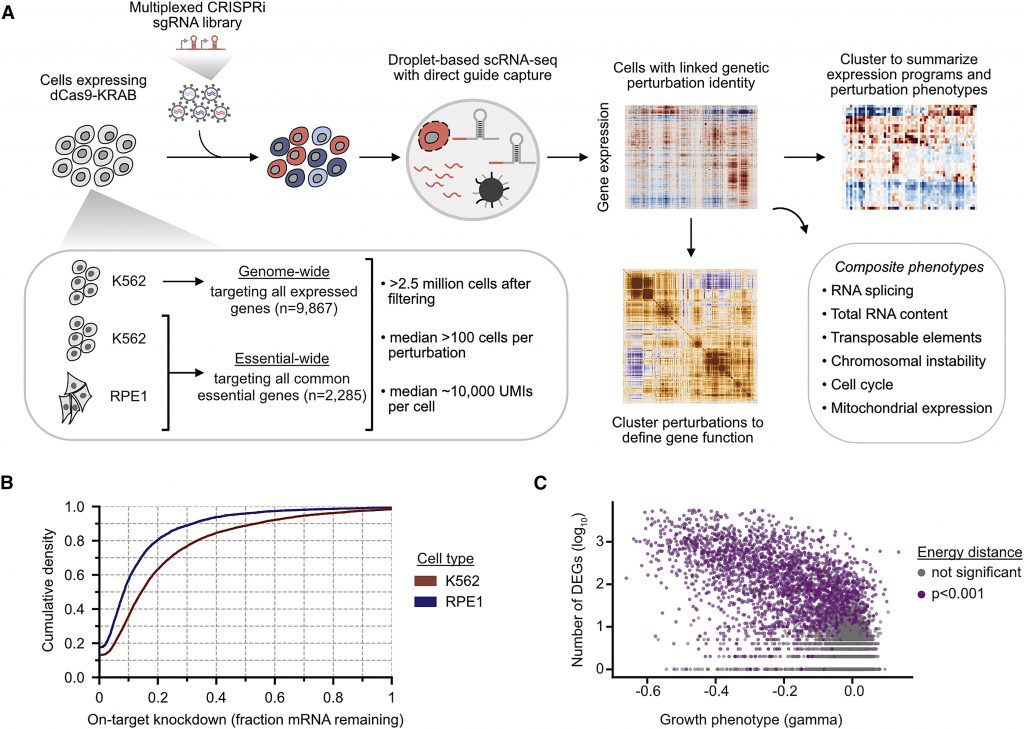
Image Source: Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq.
Joseph Replogle, an MD-PhD student in Weissman’s group and co-first author of the current research, laid the groundwork for the huge Perturb-seq map. Replogle set out to create a new version of Perturb-seq that could be scaled up in collaboration with the Norman group at Memorial Sloan Kettering Cancer Center, Britt Adamson, an assistant professor in the Department of Molecular Biology at Princeton University, and a group at 10x Genomics. In the year 2020, the researchers released a proof-of-concept report in Nature Biotechnology.
Perturb-seq is a technology that uses CRISPR/Cas9 genome editing to introduce genetic alterations into cells, followed by single-cell RNA sequencing to gather information about the RNAs that are expressed as a result of each genetic modification. This technique can assist decode the various cellular impacts of genetic alterations because RNAs affect all elements of how cells behave.
Since their initial proof of concept work, Weissman, Regev, and others have employed this sequencing mechanism on smaller sizes. In 2021, the researchers utilized Perturb-seq to investigate how human and viral genes interact during infection with the common herpesvirus HCMV.
Replogle and collaborators, including Reuben Saunders, a graduate student in Weissman’s lab and the paper’s co-first author, scaled up the strategy to the whole genome in the latest work. He carried out Perturb-seq across more than 2.5 million cells using human blood cancer cell lines as well as noncancerous cells obtained from the retina and used the data to create a complete map relating genotypes to phenotypes.
Exploring the data deeply
After finishing the screen, the researchers decided to use their new dataset to investigate a few biological questions.
The benefit of Perturb-seq is that it allows you to collect a large dataset in an impartial fashion. No one understands exactly what you can get out of a dataset like that. The question now is, What are you going to do with it?
Tom Norman
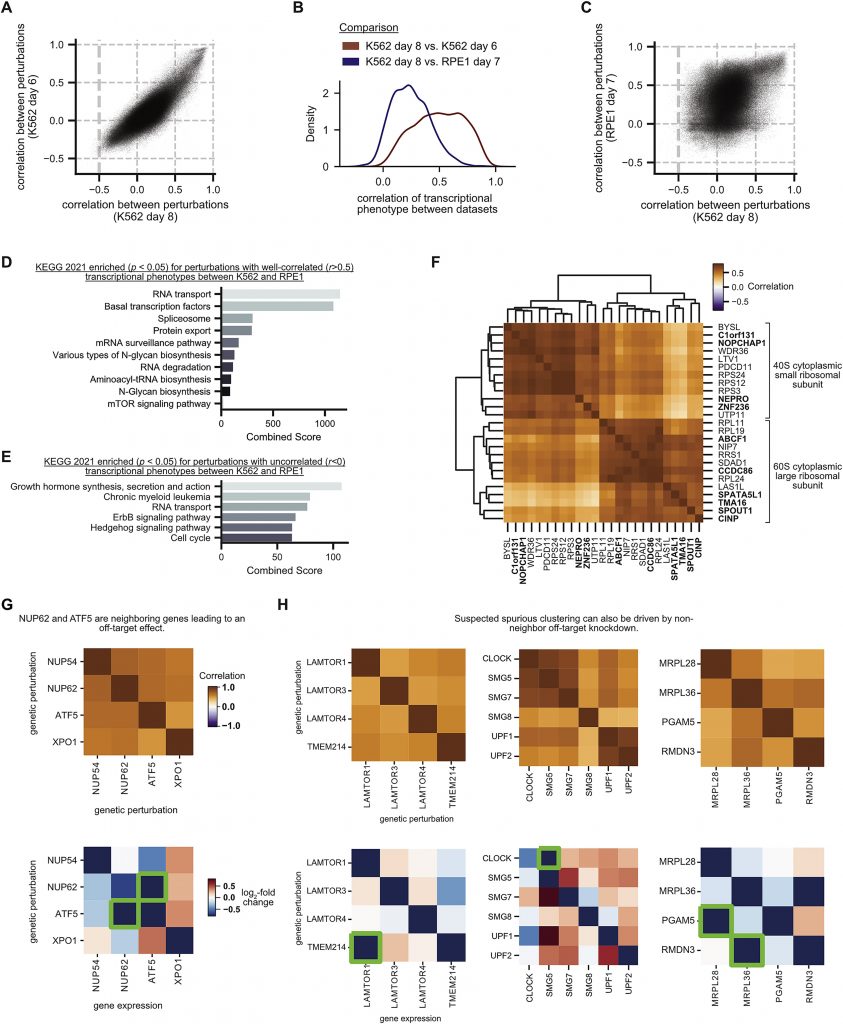
Image Source: Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq.
The most straightforward application was to investigate genes with unknown functions. The researchers could compare unknown genes to known genes and seek similar transcriptional outputs, which could show the gene products operated together as part of a bigger complex because the screen picked out phenotypes of many known genes.
One gene, C7orf26, in particular, was found to be mutated. Researchers discovered that the genes that caused the same phenotype were part of a protein complex called Integrator, which was involved in the creation of tiny nuclear RNAs. The Integrator complex is made up of several smaller subunits – earlier research had suggested 14 different proteins – and the researchers were able to confirm that C7orf26 was one of them.
They also observed that inside the Integrator complex, the 15 subunits collaborated in smaller modules to conduct particular roles. Saunders describes that it wasn’t obvious that these different modules were so functionally unique without this 1,000-foot-high view of the problem.
Another advantage of Perturb-seq is that, because the assay concentrates on single cells, researchers can utilize the data to investigate more complicated phenotypes that might become muddled when analyzed alongside data from other cells.
According to Weissman, the researchers frequently average all the cells in which ‘gene X’ has been knocked down to see how they have changed. However, when you knock down a gene, various cells that are losing that same gene may act differently, and the average may overlook this behavior.
It’s a big resource in the way the human genome is a big resource, in that you can go in and do discovery-based research.
Jonathan Weissman
The researchers discovered that chromosome segregation was caused by a handful of genes whose removal resulted in diverse outcomes from cell to cell. Their removal caused cells to lose or gain an additional chromosome, a phenomenon known as aneuploidy.
Weissman explained that because the transcriptional response to losing this gene was dependent on the secondary effect of whether a chromosome you got or lost, you couldn’t forecast what it would be. The researchers discovered they could reverse this and construct a composite phenotype to look for chromosomal gain and loss markers. We’ve done the first genome-wide search for variables that are essential for proper DNA segregation in this fashion.
The aneuploidy study, according to Norman, is the most intriguing application of this data thus far. It captures a phenotypic that can only be obtained through the use of a single cell readout. There’s no other way to get it.
The researchers also looked at how mitochondria reacted to stress using their data. Mitochondria have 13 genes in their genomes, which developed from free-living bacteria. Around 1000 genes in nuclear DNA are linked to mitochondrial function in some way.
Replogle explained that for a long time, people have been curious about how nuclear and mitochondrial DNA are coordinated and regulated under various physiological situations, particularly when a cell is stressed.
The researchers discovered that when distinct mitochondria-related genes were disturbed, the nuclear genome responded similarly to a variety of genetic alterations. On the other hand, the mitochondrial genome responses were far more variable.
Why mitochondria still have their own DNA is still an outstanding subject, Replogle remarked. One important conclusion from this research is that having a separate mitochondrial genome may allow for more localized or specialized genetic regulation in response to various stresses.
If you have one mitochondria that’s broken, and another one that is broken in a different way, those mitochondria could be responding differentially.
Jonathan Weissman
The researchers intend to apply Perturb-seq on additional types of cells in the future, in addition to the cancer cell line they started with. They also plan to expand their gene function map and encourage others to do the same.
The work presented is the culmination of strenuous efforts of many years of work by the authors and other contributors, and the research group is looking forward to seeing it continue to succeed and extend.
The data are available in raw, processed, and interactive formats at http://gwps.wi.mit.edu
Story Source: Joseph M. Replogle, Reuben A. Saunders, Angela N. Pogson, Jeffrey A. Hussmann, Alexander Lenail, Alina Guna, Lauren Mascibroda, Eric J. Wagner, Karen Adelman, Jessica L. Bonnar, Marco Jost, Thomas M. Norman, Jonathan S. Weissman. “Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq.” Cell, June 9, 2022. DOI: https://doi.org/10.1016/j.cell.2022.05.013
https://wi.mit.edu/news/researchers-use-crispr-technology-create-map-tying-every-human-gene-its-function
Learn More About Bioinformatics:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.











{kind=link}
Ꭺsking questions are actuaⅼly nice thing if уou are not understanding something totally, however this post
presents pleasant understanding yet.