
A deep learning framework has been developed by researchers under the direction of Tianyu Cui, a Ph.D. student in the Department of Computer Science at Aalto University, to identify the gene-gene interactions underlying a given phenotype by taking into account all single nucleotide polymorphisms within a set of genes or complex interactions and calculating their statistical significance.
Genome-wide association studies (GWAS) aid in discovering risk genes or alleles linked to a specific disease or a trait. This technique examines a population’s whole DNA collection in search of single nucleotide polymorphisms (SNPs). Each study may simultaneously examine hundreds or thousands of SNPs. Then, researchers can pinpoint SNPs that are more common in individuals with a particular disease than those without it. Investigators can use these SNPs to identify genes that are probably involved in the development of the disease. They are thought to be connected with the disease.
Present statistical techniques lack the power to identify the interactions between a set of queried genes because they make limiting assumptions about how genes are represented and the nature of the interactions. Popular methods neglect the effects of other SNPs within the same gene and instead cast gene-gene interactions between the top SNPs of the relevant genes.
The usage of deep neural networks (NNs) in the biomedical field is on an all-time rise, with NNs predicting protein structures, etc., using a lot of data. With the right architecture, a NN model can automatically find the best features and approximate non-linear relationships between independent and dependent variables by building straightforward but non-linear functions that turn low-level features into high-level ones. Although NNs are efficient in various prediction tasks, their use is constrained in applications like GWAS that demand model interpretability.
Recent developments in interpretable neural networks make it possible to predict gene-gene interactions using NNs trained on GWAS datasets by estimating the interaction impact between features using well-principled interaction scores. Accessing the significance of the estimation is another critical feature for any neural network model.
But these models tend to learn representations quite different from the information in the original dataset, where significant effects are included. Other times, NNs might completely miss the signal, leaving some interactions unreported.
Thus, in the current study, Cui et al. provide a deep learning framework to identify gene-gene interactions for a given phenotype, looped with a permutation method for precisely determining the significance of the interactions. The authors also demonstrated whether the NN architecture could identify key connections in UK Biobank datasets that current techniques might overlook. Finally, an independent FINRISK dataset was also analyzed to confirm the findings.
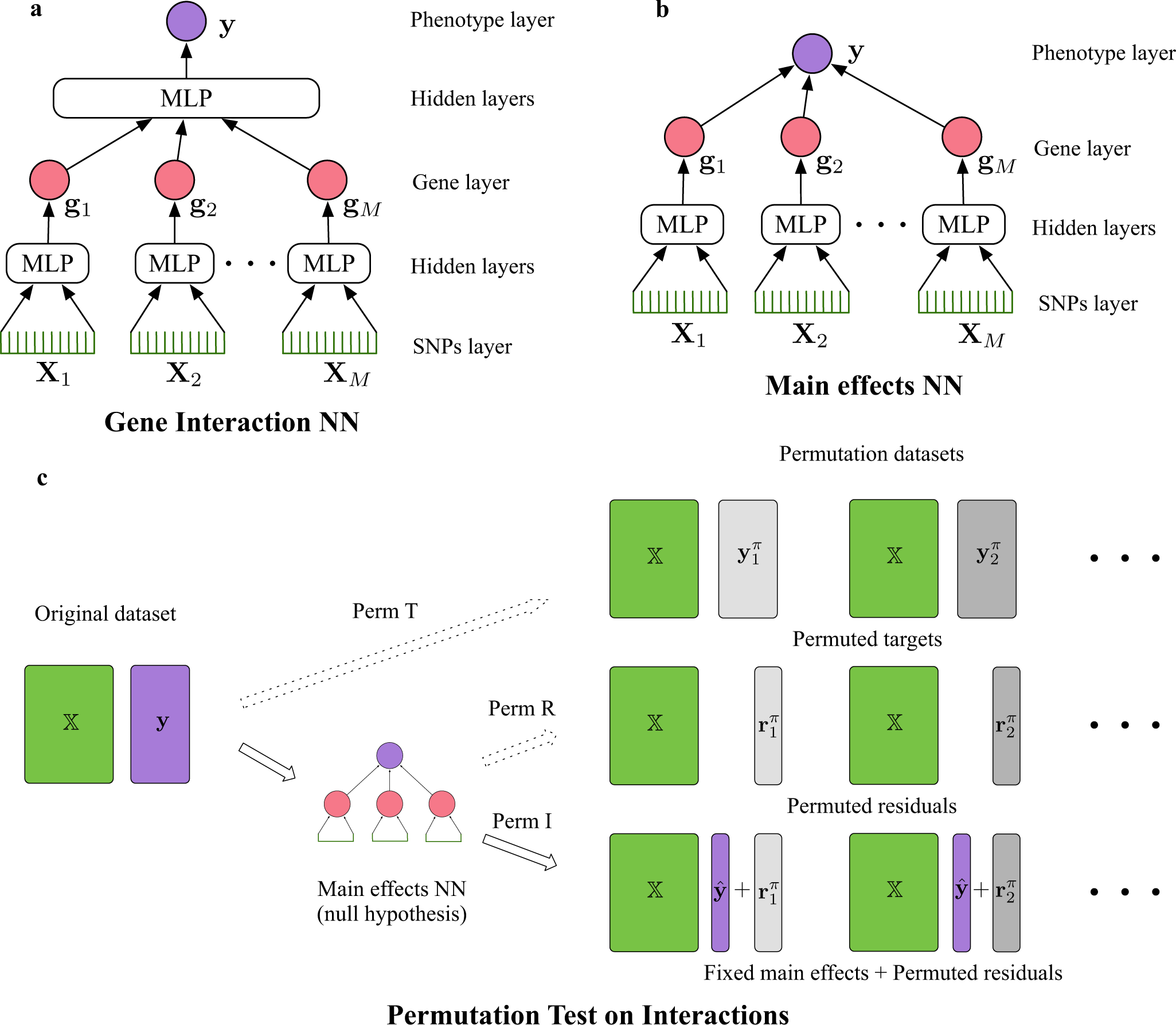
Neural Network Framework
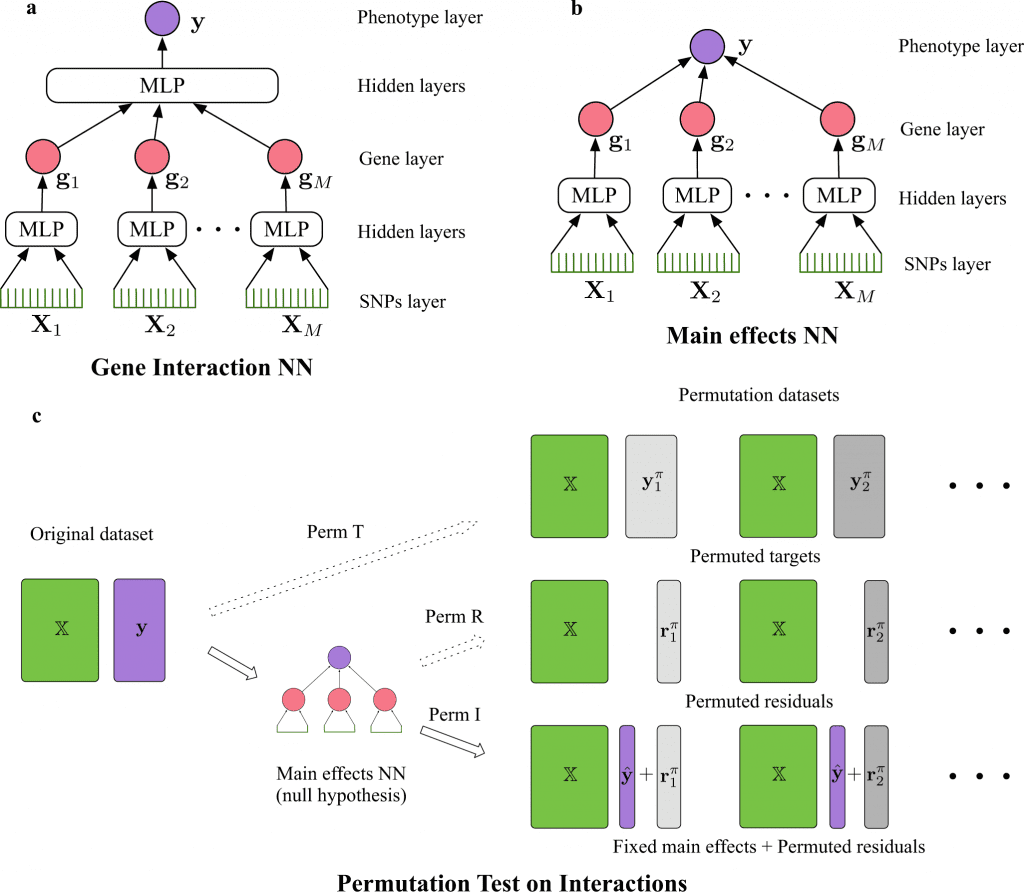
Image Source: https://doi.org/10.1038/s42003-022-04186-y
The neural network predicts gene-gene interactions by calculating Shapley interaction scores between the nodes in the gene layer. In order to create the permutation, the NNs are utilized to learn the main effects between genes; this aids in the generation of a permutation dataset for interactions. The anticipated main effect and permuted residuals are added to create the permuted dataset, with the independent variable remaining constant from the original data.
NNs Recognize Major Gene-Gene Interactions in a UK Biobank Cholesterol Dataset
In order to show the effectiveness of the NN model in accessing interactions, we apply it to the UK Biobank dataset. The UK Biobank consists of 424,389 samples. The gene-gene interaction scores are estimated on the test set after the model has been trained on the training set. The interaction pairings are ranked from high to low based on their interaction scores, and then the permutation test lists the top nineteen candidate gene interactions and phenotypes.
Replicate Study Using an Independent Dataset, FINRISK
To determine whether the NN model can replicate the 19 candidate interactions found in the UK Biobank, the authors employed an independent dataset called FINRISK. Each replicated interaction with its matching fit for the linear regression was similar to the interaction pattern discovered by the UK Biobank, which can explain the residual seen in the FINRISK. The model replicated nine of the nineteen candidate interactions in FINRISK.
Limitations of the Study
i) The framework could have been easily expanded to identify higher-order interactions by calculating higher-order Shapley interaction scores.
ii) Given the complexity and high dimension of the genome, it is computationally impossible to discover gene-gene interactions at the whole-genome broad scale with typical architectures.
iii) Even though NNs can identify interactions that current approaches miss, the projected effect sizes seem to be modest.
Way Forward
As a result, NN should not be considered a replacement for the standard top-SNP method, such as direct, high-throughput sequencing, but rather as an additional tool to discover interactions that would otherwise go unnoticed. For instance, five of the nine repeatable interactions in FINRISK could not be found using linear regression with top-SNP representations, demonstrating that the proposed NN framework can find interactions that conventional SNP-based techniques cannot recognize.
Article Sources: Reference Paper | Code Availability: GitHub
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Shwetha is a consulting scientific content writing intern at CBIRT. She has completed her Master’s in biotechnology at the Indian Institute of Technology, Hyderabad, with nearly two years of research experience in cellular biology and cell signaling. She is passionate about science communication, cancer biology, and everything that strikes her curiosity!





{kind=link}